
Spinoserebellaarinen ataksia tyyppi 5 eli SCA5
- ICD–10: G11.8, OMIM: 6000224, ORPHA: 98766
Spinoserebellaarisen ataksian tyyppi 5:n eli SCA5:n oireet rajoittuvat tavanomaisesti pikkuaivojen toimintavaurioon (Kuva 1.). SCA5 kuvattiin alun perin vuonna 1994 suuressa amerikkalaisessa perheessä, joka polveutui presidentti Abraham Lincolnin esivanhemmista. SCA5:n oireet etenevät hitaasti.
Kaikilla sairastuneilla todetaan tasapainovaikeudet, kävelyn epävarmuus sekä raajojen koordinaatio-ongelmat, epätarkkuus ja hapuilu. Lähes kaikilla on puheen sammallusta sekä useimmilla myös nystagmus. Oireiden ilmaantuminen vaihtelee, mutta tavallisimmin sairastutaan vasta aikuisiällä. SCA5 periytyy autosomisesti vallitsevasti eli dominantisti. SCA5:een liittyvä geeni on paikallistettu kromosomiin 11 ja kolme erilaista SCA5-sairauteen johtavaa geenimutaatiota on tunnistettu.
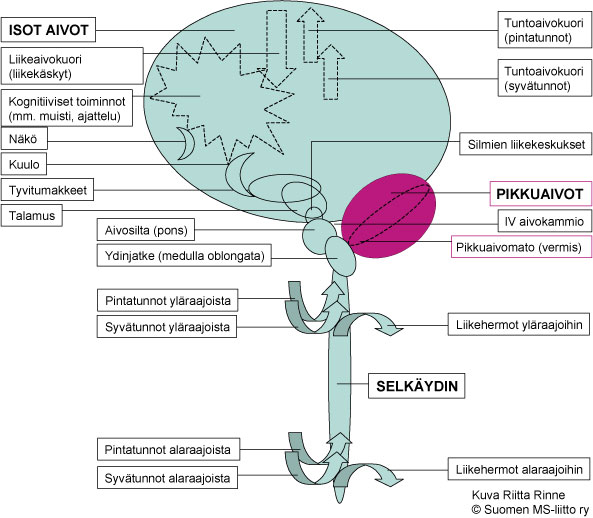
Kuva 1. SCA5:n oireet johtuvat pikkuaivojen toimintahäiriöstä.
SCA5:n oireet ovat kaikilla sairastuneilla varsin yhteneväiset ja oireiden eteneminen on hidasta. Keskimääräinen sairastumisikä on 30–40 ikävuoden välillä, mutta oireiden alkamisiässä on todettu huomattavaa vaihtelua. Nuorimmat sairastuneet ovat olleet noin 10 vuoden ikäisiä ja vanhimmat lähes 70 vuoden ikäisiä. Sairauden aiheuttama liikuntahaitta etenee siinä määrin hitaasti, että kävelyn apuvälineiden tarvetta on useimmilla vasta 20–30 vuoden sairastamisen jälkeen. Sairaus ei lyhennä elinikää. Toistaiseksi SCA5 sukuja on kuvattu kolme. Ensimmäinen suku oli amerikkalainen, seuraava suku ranskalainen ja kolmas suku saksalainen.
SCA5: taudinkuvan kuuluvat:
- tasapainon ja kävelyn epävarmuus (kävelyataksia)
- raajojen liikehäiriöt (ataksia)
- hienomotoriikan kömpelyys
- puheongelmat (dysartria)
- silmävärve (nystagmus) ja silmien liikehäiriöt
Osalla todetaan lisäksi:
- jänneheijasteiden vilkastumista
- käsien vapinaa (tremor)
- silmien liikerajoitusta sivulle katsoessa
- lihasnykäyksiä (myokymiaa) kasvoissa
- alaraajojen värinätunnon heikkenemistä
SCA5 alkaa tavallisimmin tasapainon epävarmuutena. Hienoinen tasapainohaitta voidaan havaita rappuja laskeutuessa ja horjahteluna käännöksissä. Vuosien sairastamisen jälkeen kävely muuttuu leveäraiteiseksi ja sivuaskeleita otetaan myös suoraan kävellessä. Kävelyn apuvälineet tulevat käyttöön vasta vuosikymmenienkin sairastamisen jälkeen.
Osalla potilaista ensioireena on ollut silmävärve eli nystagmus ja oireet ovat vuosien ajan saattaneet rajoittua nystagmukseen. Tyypillinen löydös on hakkaava ja lamaantumaton nystagmus. Nystagmus korostuu katseensuuntaisesti, mutta sitä todetaan myös spontaanisti. Silmien seurantaliikkeissä on hidastumista, mikä vaikeuttaa katseen kohdistamista.
Käsinäppäryys heikkenee. Pienten esineiden käsittely tuottaa ongelmia. Käsien osumatarkkuus heikkenee ja käsillä tekeminen vaatii aikaisempaa enemmän keskittymistä. Osalla potilaista on kuvattu käsivapinaa. Vapina voi ilmetä ainoastaan kohdetta hapuillessa (intention tremor) tai sitä voidaan todeta käsien ollessa levossa. Lepovapina on nopealyöntistä poiketen Parkinsonin sairauden lepovapinasta. Vapinan korostumista toiminnassa ja asennon kannattamisessa on todettu.
Puheen artikulaatio muuttuu epäselväksi, mutta pääsääntöisesti puheen ymmärrettävyys säilyy vuosikymmeniäkin jatkuneessa sairaudessa. Nielemisongelmia (dysfagia) ei ole pääsääntöisesti todettu. Ainoastaan muutamalla hyvin nuorena sairastuneella on esiintynyt dysfagiaa. Samoilla henkilöillä on todettu jänneheijasteiden vilkastumista. Näkö ja kuulo ovat säilyneet normaaleina.
SCA5:een ei liity ääreishermoston toimintahäiriöitä. Värinätunnon alenemista on todettu harvoilla, muutoin tuntotestit ovat jääneet normaaleiksi. Rakon ja suolen toimintahäiriöitä ei ole tullut esille. Potilailla ei myöskään ole havaittu muutoksia autonomisen hermoston toiminnassa. Muisti ja muut henkiset toiminnot ovat säilyneet muuttumattomina.
SCA5:n periytyminen
SCA5 periytyy autosomisesti vallitsevasti eli dominantisti (AD). Sairautta aiheuttava geeni sijaitsee kromosomissa 11q13 ja toistaiseksi on tunnistettu kolme eri pistemutaatiota SCA5-sairautta aiheuttavassa SPTBN2-geenissä. SPTBN2-geeni ohjaa spektriini-nimisen valkuaisaineen (beeta-3 spektriini) tuotantoa (Kuva 2).
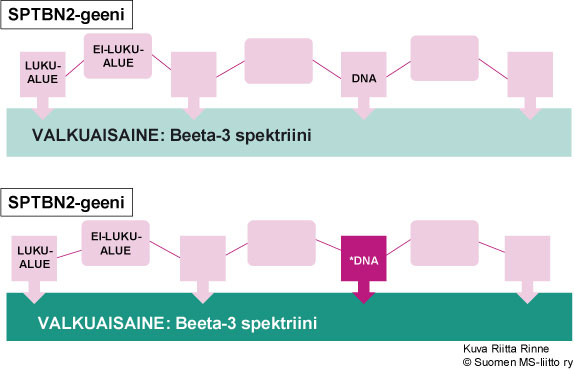
Kuva 2. SCA5-sairauden aiheuttaa SPTBN2-geenin mutaatio kromosomissa 11q13.
SCA-sairauksiin liittyy usein antisipaatio. Antisipaatiolla tarkoitetaan sairauden puhkeamista keskimäärin varhemmin laskevissa sukupolvissa. Monessa SCA-sairaudessa geenimutaatio on kolmen peräkkäisen nukleotidiemäksen monistuminen – tavallisimmin kyseessä on CAG-jakson monistuminen. Monistunut CAG-jakso on epävakaa ja sillä on taipumusta edelleen pidentyä seuraavassa sukupolvessa. SCA-sairauksien antisipaatiota selittää ainakin osittain CAG-jakson pidentymistaipumus.
SCA5:n geenimutaationa on sekä amerikkalaisessa että ranskalaisessa suvuissa toisistaan poikkeavat deleetiot (geenialueen puutokset). Saksalaisessa perheessä mutaatio on pistemutaatio (L252P). SCA5-suvuissa on osittaisesti todettu antisipaatiota: amerikkalaisissa perheissä sairauden puhkeaminen varhaistui laskevissa sukupolvissa, mutta ranskalaisessa perheessä antisipaatiota ei ole havaittu. Amerikkalaisessa perheessä seuraavan sukupolven sairastuminen tapahtui keskimäärin 15,7 vuotta aikaisemmin äidiltä periytyneessä sairaudessa ja 9,3 vuotta varhemmin sairauden periytyessä isältä. Saksalaisessa suvussa antisipaatio on keskimäärin 15,8 ± 8.0 vuotta.
Perinnöllisyysneuvonta on aiheellista järjestää aina epäiltäessä SCA-sairautta. Neuvonnassa annetaan tietoa niin sairauden laadusta kuin myös sen mahdollisesta perinnöllisyydestä sekä sairastumisen riskistä. Neuvonnassa voi keskustella myös ennakoivan geenitestin (prediktiivinen testi) mahdollisuudesta.
SCA5:n diagnostiikka ja erotusdiagnostiikka
Neurologisessa tutkimuksessa todetaan pikkuaivojen toimintahäiriöön sopivia löydöksiä: tasapainovaikeus, kävelyn ongelmat sekä käsien että jalkojen osumatarkkuuden heikkeneminen. Hienomotoriset suoritukset ovat epätarkkoja ja vastakkaiset liikesuoritukset ovat epärytmisiä. Muilta osin neurologisessa tutkimuksissa esiintyvien löydösten osalta viittaan kuvattuihin oireisiin eri perheissä. Nystagmus ja dysartria todetaan lähes kaikilla sairastuneista, mutta muiden oireiden osalta esiintymisessä on vaihtelua.
Aivojen magneettitutkimuksessa nähdään surkastumista pikkuaivomadossa (vermiksessä) ja pikkuaivokupoleissa (hemisfääreissä). Pikkuaivojen surkastumista on raportoitu jo varsin lyhyen sairastamisen jälkeen. Aivorungon rakennemuutoksia ei ole todettu. Ääreishermojen johtumishäiriöitä ei ole tullut esille. Verikokeet samoin kuin selkäydinnestenäyte ovat normaaleja. Tunnettuja SCA5-sairauteen johtavia geenimutaatioita on toistaiseksi kuvattu kolme eli amerikkalaisen, ranskalaisen ja saksalaisen perheen geenimutaatiot poikkeavat toisistaan.
Erotusdiagnostiikassa tulevat kyseeseen muut taudinkuvaltaan pikkuaivo-oireisiin rajoittuvat SCA-sairaudet (ADCA tyyppi III), mutta ranskalaisella perheellä todetut kliiniset oireet (jänneheijasteiden vilkastuminen sekä alentunut vibraatiotunto) viittaavat laajempaan keskushermoston vaurioitumiseen SCA5:ssä, joten myös ADCA I on muistettava.
SCA5:n esiintyminen
SCA5:n poikkeava geenialue on paikallistettu kromosomiin 11 ja todettiin ensimmäisenä amerikkalaisen perheen kahdessa suuressa sukuhaarassa. Tämä suku käsittää peräti 11 sukupolvea. Toinen SCA5-perhe on kuvattu Ranskassa ja toistaiseksi viimeisin SCA5-suku on kuvattu saksalaisessa perheessä vuonna 2004. Suomalaisista perheistä diagnoosia ei toistaiseksi ole tehty.
SCA5:n hoito ja kuntoutus
Sairauden hoitoon ei ole käytettävissä oireiden etenemistä hillitsevää tai sairautta parantavaa lääkehoitoa. Vitamiinilisiä on suositeltu – erityisesti silloin, kun tavanomaisesta ravinnosta niitä ei todennäköisesti riittävästi kerry. Perinnöllisyysneuvonta on oleellinen osa SCA5-sairauden hoitoa. Neuvontaa tarvitsevat sairastuneen lisäksi myös muut perheenjäsenet.
SCA5-sairauden aiheuttamat toimintarajoitteet etenevät yksilöllisesti ja toisaalta sairastumisikä vaihtelee huomattavasti. Tästä syystä kuntoutustoimet on syytä aina arvioida yksilöllisesti samoin kuin tarvittavat palvelut. Sairaus etenee huomattavan hitaasti eikä todennäköisesti lyhennä elinikää. Ammatillinen kuntoutustarve tulee arvioida tarvittaessa. Kuntoutustoimina voivat tulla kyseeseen niin kodin kuin työpaikan tehtäviä muutoksia, jotka helpottavat sairauden tuoman tasapaino- ja liikkumishaitan kanssa selviytymistä.
SCA5:n perustutkimus
Neuropatologisia tutkimusraportteja ei toistaiseksi ole.
SCA5:n geenivirhe sijaitsee kromosomissa 11q13 ja sairautta aiheuttavat mutaatiot on vastikään (2006) osoitettu SPTBN2-geenissä. SPTBN2-geeni ohjaa spektriinin tuotantoa. Elimistössä on useita spektriinejä ja SCA5-sairauteen liittyvä spektriini on nimeltään beeta-3 spektriini. Beeta-3 spektriiniä esiintyy laajalti elimistössä, mutta erityisen runsaasti sitä on pikkuaivojen Purkinje soluissa.
Beeta-3 spektriini osallistuu hermosolukalvossa tapahtuvaan viestintään tukemalla glutamaatin kuljettajaproteiinin EAAT4:n toimintaa. Glutamaatti puolestaan on yksi tärkeimmistä hermokudoksen välittäjäaineista. Soluviljelytutkimuksissa on vahvistettu virheellisen eli mutantin beeta-3 spektriinin kyvyttömyys stabiloida solukalvolla sijaitsevaa EAAT4:ää, mikä puolestaan vahingoittaa glutamaatin vaikutuksia hermosolussa.
Kirjoittaja
Riitta Rinne, LL, neurologian erikoislääkäri, Maskun neurologinen kuntoutuskeskus
Julkaistu 14.10.2002. Päivitetty 13.10.2006
- Kirjallisuusviitteet
- Ranum LPW et al., Spinocerebellar ataxia type 5 in a family descended from the grandparents of
President Lincoln maps to chromosome 11. Nature Genet 8: 280–284, 1994 - Kaakkola S ja Rinne R: Ataksiat ja niiden erotusdiagnostiikka (katsaus). Duodecim 113: 1773–1782, 1997
- Zhu S and Gerhard DS: A transcript map of an 800‐kp region on human chromosome 11q13, part
of the candidate region for SCA5 and BBS1 Hum Genet 103(6): 647–680, 1998 - Ashizawa T (Ed): Repeats may not be everything in anticipation. Neurology 53: 1164–1165, 1999
- Stevanin G et al., Clinical and MRI findings in spinocerebellar ataxia type 5. Neurology 53: 1355–1357, 1999
- Schut LJ et al., Spinocerebellar ataxia Type 5. Kirjassa: Handbook od Ataxia Disorders, ss. 435–445.
Ed. Klockgether T, Marcel Dekker Inc., New York‐Basel, 2000 - Burk K et al., Spinocerebellar ataxia type 5: clinical and molecular genetic features of a German
kindred. Neurology 62: 327–329, 2004 - Ikeda Y et al., Spectrin mutations cause spinocerebellar ataxia type 5. Nature Genet 38: 184–190,
2006 - Bauer P et al., Spectrin mutations in spinocerebellar ataxia (SCA). Bioessays 28: 785–787, 2006
- Ranum LPW et al., Spinocerebellar ataxia type 5 in a family descended from the grandparents of
Järjestötoiminta
Potilasjärjestö: Neuroliitto ry; ataksiaverkosto
Muita tietolähteitä
- The National Ataxia Foundation: www.ataxia.org
- Sveriges socialstyrelsens kunskapsdatabas om ovanliga diagnoser innehåller information om fler än 300 ovanliga sjukdomar och tillstånd.