
Spinoserebellaarinen ataksia tyyppi 1 (SCA1)
- CD–10: G11.8 Muut määritetyt perinnölliset ataksiat, OMIM: 164400, ORPHA: 98755
- Muut sairaudesta käytetyt nimet:
- Spinoserebellaarinen atrofia I
- Olivopontoserebellaarinen atrofia I (OPCA 1)
- Menzelin OPCA
Spinoserebellaarinen ataksia tyyppi 1 (SCA1) on niin pikkuaivojen ja niiden hermoratayhteyksien toimintahäiriöihin johtava etenevä neurologinen sairaus (ADCA I). Sairaus periytyy autosomisesti vallitsevasti eli dominantisti (AD). SCA1:n oireiden alkaminen ja niiden eteneminen vaihtelevat merkittävästi. Sairautta aiheuttava geenimutaatio tunnetaan ja sen määrittäminen verinäytteestä varmistaa SCA1-diagnoosin. Geenimutaation perusteella SCA1 lukeutuu polyglutamiiniataksioihin (PolyQ-ataksiat). Tavallisimmat SCA1:n erotusdiagnoosissa huomioitavat sairaudet ovat muut PolyQ-ataksiat (erityisesti SCA2 ja SCA3) sekä ne SCA-sairaudet, joiden kliiniset oireet eivät rajoitu puhtaasta pikkuaivojen vaurioitumisesta johtuviksi. SCA1 on Suomessa kolmanneksi tavallisin aikuisiän AD-periytyvä etenevä ataksiasairaus.
SCA1 alkaa tavallisimmin keski-ikäisenä (30–40 vuoden iässä), mutta sairaus voi alkaa jo huomattavasti varhemmin. Nuorimmat sairastuneet ovat olleet alle 10 vuoden ikäisiä ja vanhimmat noin 60 vuoden ikäisiä. Ensioireina todetaan tasapainovaikeus, kävelyataksia sekä puheen sammaltaminen. Pikkuaivo-oireiden lisäksi todetaan myös muita neurologisia oireita (Kuva 1). Oireet etenevät yksilöllisesti. Perheenjäsenten taudinkuvassa ja sen etenemisessä on niin ikään suuri yksilöllinen vaihtelevuus.
Aikuisiän SCA1:n taudinkuvaan kuuluvat:
- tasapainon ja kävelyn epävarmuus (kävelyataksia)
- vartaloataksia ja liikkeiden hallintavaikeus
- alaraajojen lihasjänteyden nousu (spastiteetti) ja jänneheijasteiden kiihtyminen (hyperrefleksia)
- puheen sammaltaminen ja artikulaation epäselvyys (dysartria), äänenvoimakkuuden hallitsemattomuus
- silmien liikehäiriöitä (hypermetriset sakkadit) ja silmien liikerajoituksia (oftalmoplegia, ptoosi)
Lisäksi todetaan vaihtelevasti:
- nielemisen ongelmia (dysphagia)
- jänneheijasteiden vaimeneminen tai sammuminen
- lihasten kuihtumista
- tahattomia liikehäiriöitä ja pakkoliikkeitä (dystoniaa, koreaa) sekä Parkinsonin taudin kaltaisia oireita
- tunnon alenemaa
- näköhermon pään kalpeutta ja näön heikkenemistä
- rakon toimintahäiriöitä
- painon putoamista
- kognitiivisia oireita (harvoin dementiaa)
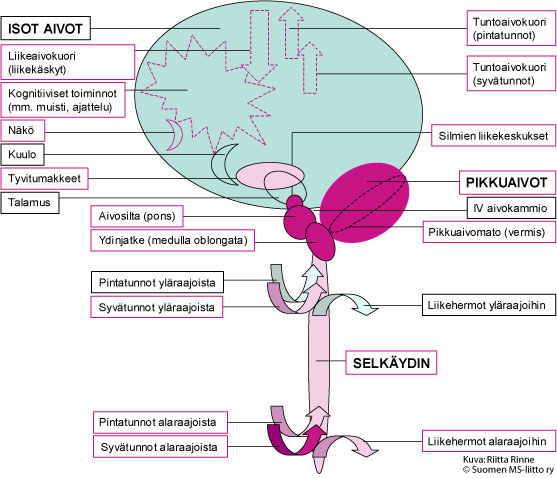
Kuva 1. Aikuisiällä alkavan SCA1:n aiheuttama neurologinen oireisto johtuu pikkuaivojen ja niiden hermoratayhteyksien toiminnan heikkenemisestä. Kuvassa on tummalla violetilla merkitty hermoston alueet, joissa SCA1-sairaus aiheuttaa aina muutoksia ja vaaleammalla violetilla ne hermoalueet, joissa muutoksia voi esiintyä.
SCA1:n taudinkuva ja oireiden eteneminen
Hienoinen tasapainohaitta ilmenee herkemmin rappuja laskeutuessa kuin niitä noustessa. Horjahtelut käännöksissä ovat tavallisia. Tasapainohaitta korostuu pimeässä tai silmien ollessa suljettuina. Vähittäisesti kävely muuttuu leveäraiteiseksi ja sivuaskeleita tulee suoraan kävellessä. Kävelyn ongelmat korostuvat hämärässä. Sairauden edetessä tasapainon ylläpitäminen on vaikeaa. SCA1:ssä todetaan myös selvä vartalon hallintavaikeus: istuma-asento horjahtaa herkästi ja kurkottelut ovat hankalia. Satunnaisesti on raportoitu voimattomuuden- ja väsymyksen tunnetta sairauden puhkeamista edeltävän vuoden aikana.
Hienomotoriikka heikkenee. Alkuun erityisesti nopeiden liikesarjojen suorittaminen sekä pienten esineiden käsittely tuottaa ongelmia. Monimutkaisten liiketaitojen – esim. pianon soittaminen, koneella kirjoittaminen jne. – opettelu ja suorittaminen on työlästä. Uusien motoristen taitojen oppiminen käy vähitellen mahdottomaksi. Käsien osumatarkkuus heikkenee ja tekeminen vaatii keskittymistä. Käsivoimat, samoin kuin jalkojenkin voimat pysyvät pitkään varsin hyvinä.
Puheen puuroutuminen ja artikulaation vaikeudet kuuluvat lähes kaikilla oirekuvaan jo sairauden ensi vuosina. SCA1-potilaalla esiintyy usein ongelmia nielemisessä: ruoka ja erityisesti juomat menevät väärään kurkkuun. Nielemisongelmia lisää oirekuvaan liittyvä yskimisrefleksin heikkeneminen. Nielemiseen liittyvät ongelmat voivat olla yksi hankalimmista ja henkeä uhkaavimmista oireista pitkään jatkuneessa sairaudessa. Sairauteen liittyy myös painon putoamista, joka ei välttämättä ole yhteydessä nielemisen ongelmiin. Painon putoaminen alkaa sairauden keskivaiheessa ja siihen on vaikea vaikuttaa ruokavaliolla tai lääkehoidoin.
Lähes kaikilla todetaan silmien liikehäiriöitä sekä sairauden edetessä myös silmien liikerajoituksia; yläluomien roikkumista eli ptoosia on harvemmin (40 %:lla). Sairauden ensivuosina silmien seurantaliikkeet ovat ”vilkastuneet” (hypermetriset sakkadit), sairauden edetessä seurantaliikkeet voivat hidastua (hypometriset sakkadit). Joskus todetaan näön heikkenemistä, mikä on todennäköisempää varhain sairastuneilla. Silmänvärvettä (nystagmusta) esiintyy harvoin (1/10). Kasvohermojen heikkoutta (facialispareesi) todetaan 40 %:lla.
Värinätunnon heikkenemistä ja nivelten asentotunnon huononemista todetaan noin 80 %:lla. Yli puolella sairastuneista esiintyy muita ääreishermoston toimintahäiriöitä. Pakkoliikkeet (dystonia, korea, myoklonia) ja liikkeiden yleinen hidastuminen (bradykinesia) kuuluvat tavanomaisesti pitkään jatkuneeseen sairauteen.
Sairauteen saattaa liittyä rakon pidätyskyvyn ongelmia (20–50 %:lla). Joka toisella SCA1-potilaista on nukahtamisen vaikeutta sekä alaraajojen levottomuutta, erityisesti levossa.
Pääsääntöisesti SCA1 ei aiheuta muutoksia, jotka vaikeuttaisivat sairastavan kykyä päättää ja huolehtia omista asioistaan. SCA1 ei myöskään aiheuta muutoksia puheen sisältöön, sanojen muistamiseen tai puheen ymmärtämiseen. Tunnereaktioiden eli emootioiden säätelyhäiriötä on todettu osalla potilaista sairauden edetessä, samoin ärsykeherkkyyden lisääntymistä (impulssien kontrolliongelmat). Sairauteen liittyy myös kognitiivisia muutoksia. Ongelmia on todettu toimintojen aloittamisessa ja suorittamisessa, keskittymisessä sekä kielellisessä muistissa. Näönvarainen muisti pysyy yleensä normaalina. Laaja-alaisia, häiritsevästi arkipäivän toimintaan vaikuttavia kognitiivisia muutoksia on todettu SCA1:n yhteydessä harvalla ja yleensä vasta sairauden muutoinkin aiheuttaessa huomattavia toimintarajoitteita.
Liikunnalliset ongelmat kehittyvät keskimäärin 5–10 vuoden kuluessa siinä määrin hankaliksi, että apuvälineet ovat välttämättömiä. SCA1 lyhentää useimpien kohdalla elinikää ja oireiden kesto on keskimäärin 10–15 vuotta (10–28 vuotta). Eräässä suuressa eurooppalaisessa suvussa kuoliniäksi todettiin 54,1 ± 6,5 vuotta. SCA1-potilaiden välittömistä kuolinsyistä on vähän julkaistua tietoa. Keuhkokuume (aspiraatio pneumonia), ilmarinta sekä vaikea kuumeinen infektio (sepsis) on raportoitu.
SCA1:n periytyminen
SCA1 periytyy autosomisesti vallitsevasti eli autosomissa dominantisti (AD). Sairautta aiheuttava geenivirhe tunnistettiin kromosomissa 6(p23) vuonna 1993. Geenitesti varmistaa SCA1:n diagnoosin. SCA1-sairauden geeni on ATXN1 ja sen tuottama valkuaisaine on Ataksiini-1. SCA1-sairaudessa geenimutaatio on geenin luentakehyksen sisäinen ylipitkä, epävakaa CAG-jakso. Normaalissa ATXN1-geenissä on 6–36 CAG-jaksoa. SCA1-sairauden geenissä on yli 39 CAG-jaksoa. CAG-nukleotidikomikko ohjaa aminohappo glutamiinin syntymisen. Seurauksena on ylipitkä glutamiiniketju (PolyQ-ketju) Ataksiini-1:ssä (Kuva2.).
Ylipitkän CAG-jakson epävakaus (dynaaminen mutaatio) merkitsee CAG-jakson taipumusta pidentyä seuraavassa sukupolvessa. CAG-jakson pidentymistaipumus on havaittu etenkin sairauden periytyessä isältä. CAG-jakson pituuden ja oireiden puhkeamisen välillä on todettu käänteistä korrelaatiota: pitkä CAG-jakso on yhteydessä oireiden varhaiseen puhkeamiseen ja nopeaan taudinkulkuun (genotyyppi – fenotyyppi korrelaatio). Sairauden varhaistumista seuraavassa sukupolvessa kutsutaan antisipaatioksi. SCA1-sairaudessa antisipaatio vaihtelee (km. 5–10 v). CAG-jakson pituuden perusteella ei oireiden puhkeamisikää tai niiden etenemistä voi ennustaa. On todettu, että sisarussarjassa samanpituisen CAG-jakson omaavat voivat sairastua eri-ikäisinä, eivätkä sisarusten oireet etene samaan tahtiin. CAG-jakson pituus ei siis yksinomaisesti määrää SCA1-sairauden oirekulkua.
SCA1:n diagnoosi varmistuu DNA-testillä
DNA-testi varmistaa SCA1:n diagnoosin. DNA-testiä voidaan käyttää myös sairautta ennakoivana (ennustava eli prediktiivinen geenitesti). Prediktiivinen geenitesti voidaan tehdä halutessa täysi-ikäisiltä oireettomilta, riskissä olevilta sukulaisilta tai sikiönäytteestä.
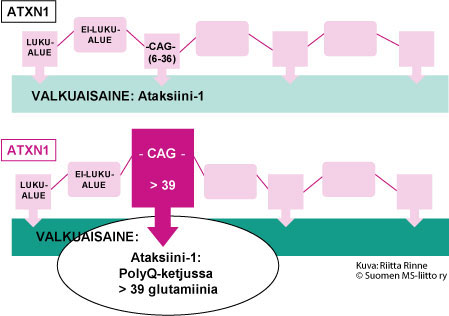
Kuva 2. SCA1-sairaudessa geenivirhe aiheuttaa Ataksiinii-1 valkuaisaineeseen ylipitkän PolyQ-ketjun syntymisen.
SCA1:n diagnoosi ja erotusdiagnoosi
SCA1-sairauden diagnoosiin ja erotusdiagnoosiin tarvitaan neurologinen tutkimus sekä sen perusteella arvioitavat laboratorio-, kuvantamis- ja muut hermotoimintojen tutkimukset. Muiden erikoisalojen konsultaatiot ovat usein perusteltuja. Perinnöllisyyslääkärin konsultaatio ja neuvonta sairastuneelle ja hänen sukulaisille on aina tarpeen.
Laboratoriotutkimukset verinäytteestä, lukuun ottamatta diagnostista DNA-testiä, ovat normaalit, samoin selkäydinnestenäyte (likvor-tutkimus). Magneettitutkimuksessa todetaan hermokudoskatoa etenkin pikkuaivojen, aivorungon (olivopontoserebellaarinen atrofia) ja vähäisemmässä määrin selkäytimen alueella. Neurofysiologisissa tutkimuksissa motoristen herätepotentiaalien ja liikehermojen johtumisajat ovat pidentyneet. Tuntohermojen vaurion osoittaminen ENMG-tutkimuksessa on harvinaisempaa. Aivojen aineenvaihduntaa kuvaavassa positroni emissio tomografiassa (PET-tutkimus) on todettu alentunutta sokeriaineenvaihduntaa pikkuaivoissa ja aivorungossa.
Erotusdiagnoosissa tärkein ryhmä on muut PolyQ-ataksiat (erityisesti SCA2 ja SCA3) . Periaatteessa myös muut SCA-sairaudet, joiden oireet eivät rajoitu puhtaasti pikkuaivo-oireisiin (ADCA I), tulevat kyseeseen. Nuorena alkavan SCA1:n erotusdiagnostiikkaan kuuluu SCA7. Perinnöllinen, nuorella iällä alkava Parkinsonin tauti sekä myös etenevän oirekuvan mukainen MS-tauti (primaaristi progressiivinen eli PP-MS) voivat alkaa samanikäisenä ja samankaltaisin oirein. SCA1-diagnoosi on satunnaisesti todettu myös ataksiapotilailla, joiden suvussa ei vastaavasti sairastuneita ole aikaisemmastaan diagnosoitu (sporadinen).
SCA1:n esiintyminen
SCA1-diagnooseja Suomessa on tehty pohjois- ja itäsuomalaisilla suvuilla. SCA1 on kolmanneksi yleisin (4%) AD-periytyvistä ataksiasairauksista Suomessa. SCA1 on tavallinen AD-ataksia Etelä-Afrikassa – 40,5% määritetyistä SCA.sairauksista – sekä suhteellisen tavallinen myös Englannissa, Italiassa, Serbiassa, Australiassa sekä Intiassa, joissa se edustaa noin 15–25 % AD-periytyvistä ataksioista.
SCA1:n aiheuttamat hermovauriot
SCA1:n neuropatologiset löydökset paikallistuvat pikkuaivojen, ponsin, oliva-tumakkeiden, alempien aivohermotumakkeiden, selkäytimen takajuosteen ja spinoserebellaaristen ratojen alueille. Alueilla todetaan hermosolukatoa (atrofia). Huomattavan voimakasta atrofiaa on todettu dentatorubraalisissa hermoradoissa. Pikkuaivoissa todetaan Purkinjen solujen ja olivoserebellaaristen ratayhteyksien atrofiaa. Selkäytimessä atrofiaa todetaan niin spinoserebellaarisissa radoissa kuin Clarkin pylväikössä.
SCA1:n hoito ja kuntoutus
Ataksian lievittäminen käytössä olevin lääkkein ei onnistu. Vapinaoireeseen voi yrittää beeta-salpaajaa. Lisääntynyttä lihastonusta vähentävät spastisiteetin hoitoon tarkoitetut lääkkeet. Parkinsonin taudin lääkehoidossa käytettävät L-dopamiini tai dopamiinin vaikutusta tehostavat valmisteet (dopamiini-agonistit) auttavat usein levottomiin jalkoihin. Rakon toimintahäiriöihin on niin ikään saatavissa apua. Aloitettavan oirelääkkeen tehon arvioinnissa on oltava kriittinen. Etenevässä sairaudessa lääkkeen tarpeellisuutta ja sen annosta on syytä tarkistaa. Hoitava lääkäri antaa ohjeet annosmäärän säätelyyn. Pitkään käytössä olleiden keskushermostoon vaikuttavien lääkkeiden lopettamista ei saa tehdä äkillisesti.
Kuntoutustarpeen arvioiminen ja kirjaaminen kuntoutussuunnitelman ja seuranta ovat keskeinen osa SCA1-sairauden hyvää hoitoa. Kuntoutussuunnitelmaan on kirjattava eri terapioiden tarve, tarvittavat apuvälineet ja muut selviytymistä tukevat keinot. Tarvittavat palvelut kirjataan palvelusuunnitelmaan.
Liikuntakyvyn säilymistä tuetaan fysioterapialla. Tasapainon, lihasten hallinnan sekä koordinaation harjoittaminen omaehtoisesti kannattaa. Soveltuvista liikunnan muodoista saa ohjeita fysioterapeutilta, samoin kuin opastusta lihashuollosta ja rentoutuksesta. Puheterapeutin arviot ja ohjeet ylläpitävät kommunikaatiotaitoja.
SCA1:een liittyy usein nielemisongelmia. Ruoka nielaistaan ”väärään kurkkuun” eli henkitorveen (aspiraatio). Aspiraatio altistaa toistuville keuhkotulehduksille ja pahimmillaan tukehtumiselle. Aspiraation riski tutkitaan videofluorometrialla. Nielemisen turvallisuuteen voidaan vaikuttaa. Nielemisen koordinointiongelmissa on vältettävä karkeusasteeltaan vaihtelevia ruokia. Kiinteä ruoka ja nesteet on nautittava erikseen. Nielemisen turvallisuutta lisäävät ruuan ja nesteiden tasalaatuisuus ja ruokailun rauhoittaminen. Vatsanpeitteiden läpi asetettavaa PEG-letkua suositellaan silloin, jos ruokailu aspiraatiovaaran takia ei ole turvallista.
Niin kommunikaatiota, arkipäivän toimien suorittamista kuin myös liikkumista on mahdollista helpottaa apuvälinein. Sairauden aiheuttaessa lisääntyvää avun- ja hoivan tarvetta on tehtävä palvelusuunnitelma. Kuntoutussuunnitelman tavoin myös palvelusuunnitelman ajantasaisuus on sovitusti tarkistettava.
SCA1:n perustutkimuksesta
SCA1-sairauden tutkimukseen on kehitetty niin kärpäs- kuin hiirimalleja. Ataksiini-1:tä esiintyy myös muissa soluissa kuin hermosoluissa. Runsaasti Ataksiini-1:tä on pikkuaivojen Purkinjen soluissa. Ataksiini-1-valkuaisaineen sijaitsee solussa tuman sisällä. Ylipitkän PolyQ-ketjun sisältävän Ataksiini-1:tä on sekä tumissa että solulimassa. Poikkeavan Ataksiini-1:n kertyminen soluun on tunnusomaista SCA1-sairaudessa. Tuma vastaa solun uusiutumisesta. Ataksiini-1 sitoutuu tumassa kromosomeihin ja heikentää mm. RNA-DNA:n luentaa. Solumekanismit eivät välttämättä riitä hajottamaan syntynyttä ylipitkän PolyQ-ketjun omaavaa Ataksiin-1:tä. Patologinen Ataksiini-1 sinänsä estänee solun proteasomin (multikatalyyttisiä proteaaseja) toimintaa. Proteasomit vastaavat soluissa tiettyjen valkuaisaineiden hajoittamisesta. Koe-eläintutkimuksissa on myös kyetty estämään patologisen Ataksiini 1:n tuotantoa solussa, mikä sinänsä antaa uutta pontta sairauden hoitomahdollisuuksille tulevaisuudessa.
Kirjoittaja
Riitta Rinne, LL, neurologian erikoislääkäri
Maskun neurologinen kuntoutuskeskus
Julkaistu 14.10.2002
Päivitetty 16.6.2006
- Kirjallisuusviitteet
- Orr HT et al: Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1.
Nat Genet 4: 221 – 226, 1993 - Giunti P et al.: The trinucleotide repeat expansion on chromosome 6p (SCA1) in autosomal
dominant cerebellar ataxias. Brain 117: 645 – 649, 1994 - Genis D et al., Clinical, neuropathologic, and genetic studies of a large spinocerebellar ataxia type
1 (SCA1) kindred: (CAG)n expansion and early premonitory signs and symptoms. Neurology 45: 24
– 30, 1995 - Schöls L et al.: Spinocerebellar ataxia type 1: clinical and neurophysiological characteristics in
German kinreds. Acta Neurol Scand 92: 478 – 485, 1995 - Bürk K et al.: Autosomal dominant cerebellar ataxia type I. Clinical features and MRI in families
with SCA1, SCA2 and SCA3. Brain 119: 1497 – 1505, 1996 - Kaakkola S ja Rinne R: Ataksiat ja niiden erotusdiagnostiikka (katsaus). Duodecim 113: 1773 –
1782, 1997 - Cummings CJ et al.: Chaberone suppression of aggregation and altered subcellular proteasome
localization imply protein misfolding in SCA1. Nature Genetics 19: 148 – 153, 1998 - Grewal RP et al.: Clinical and genetic analysis of a distinct autosomal dominant spinocerebellar
ataxia. Neurology 51: 1423 – 1426, 1998 - Moseley ML et al.: Incidence of dominant spinocerebellar and Friedreich triplet repeats among
361 ataxia families. Neurology 51: 1666 – 1671, 1998 - Rivaud‐Pechoux S et al.: Eye movement abnormalities correlate with genotype in autosomal
dominant cerebellar ataxia type I. Ann Neurol 43: 297 – 302, 1998 - Schöls L et al.: Sleep disturbance in spinocerebellar ataxias. Is the SCA3 mutation a cause of
restless legs syndrome? Neurology 51: 1603 – 1607, 1998 - Trojano l et al., Determinants of cognitive disorders in autosomal dominant cerebellar ataxia type
1. J Neurol Sci 157: 162 – 167, 1998 - Vig PJS et al: Reduced immunoreactivity to calcium‐binding proteins in Purkinje cells precedes
onset of ataxia in spinocerebellar ataxia‐1 transgenic mice. Neurology 50: 106 – 113, 1998 - Duyckaerts C et al., Nuclear inclusion in spinocerebellar ataxia type 1. Acta Neuropathol (Berl.) 97:
201 – 207, 1999 - Orr T and Klockgether T: Spinocerebellar Ataxia 1. Kirjassa: Handbook of Ataxia Disorders, 343 ‐
361. Toim. Klockgether T, Marcel Dekker Inc., New York‐Basel, 2000 - Storey E et al., Frequency of spinocerebellar ataxia types 1, 2, 3, 6, and 7 in Australian patients
with spinocerebellar ataxia. Am J Med Genet 95: 351‐357, 2000 - Srivastava AK et al., Molecular and clinical correlation in five Indian families with spinocerebellar
ataxia 12. Ann Neurol 50: 796‐800, 2001 - Bryer A et al., The hereditary adult‐onset ataxias in South Africa. J Neurol Sci 216: 47‐54, 2003
- Bürk K et al., Cognitive deficits in spinocerebellar ataxia type 1, 2, and 3. J Neurol 250: 207 – 211,
2003 - Emamiam ES et al., Serine 776 of ataxin‐1 is critical for polyglutamine‐induced disease in SCA1
transgenic mice. Neuron 38: 375 – 387, 2003 - Nagaoka U et al., Attenuated nuclear shrinkage in neurones with nuclear inclusions of SCA1 brains.
J Neurol Neurosurg Psychiatry 74: 797 – 6001, 2003 - Brusco A et al., Molecular genetics of hereditary spinocerebellar ataxia. Mutation analysis of
spinocerebellar ataxia genes and CAG/CTG repeat expansion detection in 225 Italian families. Arch
Neurol 61: 727 – 733, 2004 - Guerrini L et al., Brainstem neurodegeneration correlates with clinical dysfunction in SCA1 but not
in SCA2. A quantitative volumetric, diffusion and proton spectroscopy MR study. Brain 127: 1785 –
1795, 2004 - Park Y et al., Proteasome function is inhibited by polyglutamine‐expanded Ataxin‐1, the SCA1 gene
product. Mol Cell 19, 23 – 30, 2004 - Serra HG et al., Gene profiling links SCA1 pathophysiology to glutamate signalling in Purkinje cells
of transgenic mice. Hum Mol Genet 13: 2535 – 2543, 2004 - Tsai C‐C et al., Ataxin 1, a SCA1 neurodegenerative disorder protein, is functionally linked to the
silencing mediator of retinoid and thyroid hormone receptor. PNAS 1010, 4047 – 4052, 2004
van de Warrenburg BPC et al., Peripheral nerve involvement in spinocerebellar ataxias. Arch
Neurol 61: 257 – 261, 2004 - Xia H et al., RNAi suppresses polyglutamine‐induced neurodegeneration in a model of
spinocerebellar ataxia. Nat Med 10: 816 – 820, 2004 - Zu T et al., Recovery from polyglutamine‐induced neurodegeneration in conditional SCA1
transgenic mice. J Neurosci 24: 8853 – 8861, 2004 - Gerwig M et al., Timing of conditioned eyeblink responses is impaired in cerebellar patients. J
Neurosci 25: 3919 – 3931, 2005 - Irwin S et al., RNA association and nucleocytoplasmic shuttling by ataxin‐1. J Cell Sci 118: 233 –
242, 2005 - Juvonen V et al. The occurence of dominant spinocerebellar ataxias among 251 Finnish ataxia
patients and the role of predisponding large normal alleles in a genetically isolated population.
Acta Neurol Scand 111: 154 – 1562, 2005 - Kaytor MD et al., A cell‐based screen for modulators of ataxin‐1 phosphorylation. Hum Mol Genet
14: 1095 – 1105, 2005 - Rakowicz M et al., Spinocerebellar ataxias type 1 and 2: comparison of clinical, electrophysiological
and magnetic resonance evaluation (artikkeli puolaksi). Neurol Neurochir Pol 39: 263 – 275, 2005 - Tanaka M et al., A novel therapeutic strategy for polyglutamine diseases by stabilizing aggregation‐
prone proteins with small molecules. J Mol Med 83: 343 – 352, 2005 - van de Warrenburg BPC et al., Age at onset variance analysis in spinocerebellar ataxias: a study in
a Dutch‐Frech cohort. Ann Neurol 57: 505 – 512, 2005 - Wüllner U et al., Dopamine transporter positron emission tomography in spinocerebellar ataxias
type 1, 2, 3 and 6. Arch Neurol 62: 1280 – 1285, 2005 - Dragasevic NT et al., Frequency analysis and clinical characterization of different types of
spinocerebellar ataxia in Serbian patients. Mov Disord 21(2): 187 – 191, 2006
- Orr HT et al: Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1.
Järjestötoiminta
Potilasjärjestö: Neuroliitto ry; ataksiaverkosto
Muita tietolähteitä
- The National Ataxia Foundation: www.ataxia.org
- Sveriges socialstyrelsens kunskapsdatabas om ovanliga diagnoser innehåller information om fler än 300 ovanliga sjukdomar och tillstånd.
- European Clinical Trials Information Network kerää ja julkaisee sivustollaan tietoa SCA1:n liittyvistä kliinisistä tutkimuksista (clinicaltrials.eu).