
Spinoserebellaarinen ataksia tyyppi 8 (SCA8)
- ICD-10: G11.8, OMIM: 603680, ORPHA: 98760
Spinoserebellaarinen ataksia tyyppi 8 (SCA8) on pikkuaivojen ja niiden hermoratayhteyksien toimintahäiriöstä johtuva sairaus.
SCA8:n oireet rajoittuvat usein puhtaaseen pikkuaivovaurioon, minkä seurauksena potilailla on tasapainon ja kävelyn ongelmia sekä puheartikulaation hidastumista. Osalla SCA8:ssa todetaan kognitiivisia tai psyykkisiä muutoksia. SCA8 periytyy periytyy autosomisesti vallitsevasti eli dominantisti. Sairautta aiheuttava geenimutaatio tunnetaan. SCA8 on Suomessa tavallisin aikuisiällä alkava SCA-sairaus.
SCA8 alkaa tyypillisesti vasta lähellä 40 ikävuotta (km. 39 vuoden iässä). Italialaisessa tutkimuksessa oireiden alkaminen oli vasta yli 50 vuoden ikäisenä. Vaihtelu oireiden alkamisessa on suuri (1–65). Lähes poikkeuksetta sairaus on hidaskulkuinen eikä vaikuta elinennusteeseen. Miesten ja naisten oireiden puhkeamisiässä ei ole eroa, mutta perheenjäsenten oireiden etenemisessä todetaan vaihtelua. Liikunnan apuvälineet tulevat tarpeellisiksi runsaan 10–20 sairastamisvuoden jälkeen, osalla sairastamisaika ennen apuvälineiden käyttöönottoa voi olla huomattavastikin pidempi. Puheen hidasrytmisyys, kangertaminen on tyypillistä ja usein haittaavin SCA8:n oire.
SCA8:n taudinkuva
SCA8:aan liittyvät aina tasapainovaikeudet, kävelyn epävarmuus sekä raajojen koordinaatio-ongelmat, epätarkkuus ja hapuilu. Sairastuminen tapahtuu usein vasta keski-ikäisenä, mutta sairastumisiässä on suurta yksilöllistä vaihtelua. Vaihtelua todetaan myös perheenjäsenten oireiden etenemisnopeudessa. Oireet rajoittuvat pikkuaivoperäisiin oireisiin ja ovat luonteeltaan eteneviä (Kuva 1).
Aikuisiän SCA8:n taudinkuvaan kuuluvat:
- tasapainon ja kävelyn epävarmuus (kävelyataksia)
- puheen kankeus, hitaus, sammallus ja artikulaation epäselvyys (dysartria)
- nielemisen ongelmat (dysfagia)
- raajojen liikkeiden holtittomuus (ataksia)
- vartalon hallintavaikeudet (vartaloataksia)
- silmien liikehäiriöt
Osalla todetaan lisäksi:
- silmänvärve (nystagmus)
- alentunut värinätunto
- jänneheijasteiden joko vilkastumista tai vaimentumista
- vapinaoiretta (tremor)
- neuropsykologisia oireita
Lähes kaikilla todetaan ensioireina tasapainoepävarmuus ja liikkumisen vaikeudet, mutta joillakin sairauden ensioireena voi olla puheen kangertaminen. Sairastamisen ensivuosina tasapainovaikeudet voivat olla varsin vähäisiä ja ainoastaan vaativimmissa liikunnallisissa tilanteissa ilmeneviä. Hienoinen tasapainohaitta voidaan havaita rappuja laskeutuessa tai horjahteluna käännöksissä. Vähittäisesti kävely muuttuu leveäraiteiseksi ja sivuaskeleita otetaan myös suoraan kävellessä. SCA8:ssa kaikilla todetaan vartaloataksiaa: istuma-asento horjahtaa ja kurkottelut ovat hankalia.
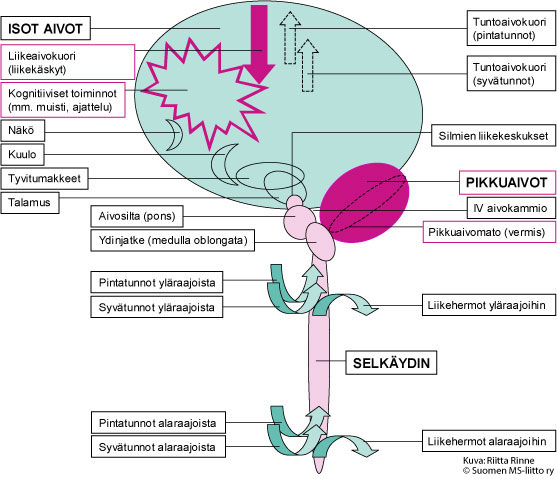
Kuva1. SCA8-sairaudessa todetaan pikkuaivovaurioon sopiva kliininen oireisto. Vaihtelevasti todetaan ylemmän liikehermon vaurioita sekä kognitiivisia muutoksia.
Käsinäppäryys hidastuu. Tarkkuutta vaativat tehtävät sekä pienten esineiden käsittely tuottavat ongelmia. Käsien osumatarkkuus heikkenee ja käsillä tekeminen vaatii keskittymistä. Käsien vapina (aktion tremor) haittaa tarkkaa työskentelyä. Käsivoimat, samoin kuin jalkojenkin voimat ovat pääsääntöisesti hyvät.
Puheen hitaus ja artikulaation ongelmat kuuluvat aina oirekuvaan. Dysartria todetaan joillakin lähes samanaikaisesti tasapainovaikeuksien kanssa. Sairauden ensivuosina todetaan myös silmien liikehäiriöitä, lähinnä katseen suuntaamisen ongelmia. Horisontaalista nystagmusta todetaan 2/3:lla sairastavista.
Joillakin todetaan muutoksia jänneheijasteissa. Etenkin alaraajojen lihaksissa voi olla lihasten jänteyden nousua (spastisiteettia) ja jänneheijasteiden vilkastumista. Satunnaisesti on todettu myös vaimentuneet jänneheijasteet. Pakkoliikkeitä ei ole yhdistetty SCA8:aan.
Neuropsykologisia häiriöitä voi liittyä oirekuvaan. Kognitiiviset muutokset ovat pääsääntöisesti lieviä ja aiheuttavat ongelmia toimintojen suunnittelussa, keskittymisessä, tarkkaavaisuudessa sekä myös kielellisessä ilmaisussa. Joidenkin kohdalla neuropsykologiset muutokset ovat kehittyneet arkipäivän toimintoja haittaaviksi (dementia). SCA8:ssa on raportoitu mielialaan liittyviä muutoksia (depressiota, bipolaarista mielialahäiriötä sekä rajatilaa).
Pitkään jatkuneeseen sairauteen voi liittyä myös nielemisen ongelmia samoin kuin silmien liikkeiden rajoittumista. Yskimisrefleksi voi olla heikentynyt. Puheen sisältöön tai sen ymmärtämiseen sairaus ei vaikuta. Tuntomuutokset rajoittuvat yleensä värinätunnon alenemiseen. Rakon- tai suolen toimintahäiriöitä ei ole raportoitu.
SCA8:n periytyminen
SCA8 periytyy autosomisesti vallitsevasti eli dominantisti. Geenitesti varmistaa SCA8 diagnoosin. Testi tehdään verinäytteestä. SCA8:n geenimutaatio sijaitsee kromosomissa 13 alueella q21 geenin 3´-päässä. Normaalisti SCA8-geenissä geenin vastinpareissa (alleeleissa) CTG-jakson pituus on 16–34. Sairastuneiden CTG-jakso on pidentynyt ollen tavallisimmin >100 CTG-jaksoa (Kuva 2.)
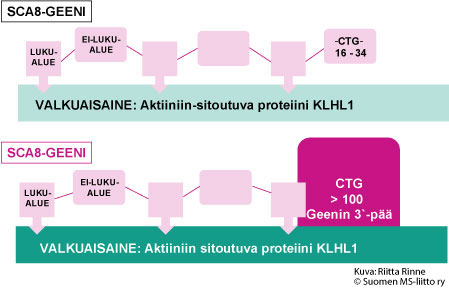
Kuva 2. SCA8-geeni ohjaa Aktiiniin-sitoutuvan valkuaisaineen syntymistä. Proteiini on nimetty Kelch-like1-proteiiniksi (KLHL1). KLHL1 proteiinia on hermosoluissa, erityisesti keskushermostossa.
SCA-sairauksissa, joissa geenimutaatio on ylipitkä nukleotidijakso (PolyQ-ataksiat), todetaan usein käänteinen korrelaatio nukleotijakson pituuden ja sairauden alamisen välillä. SCA8:ssa kiistatonta genotyypin määräämää käänteistä korrelaatiota ei ole voitu todeta kaikissa tutkituissa perheissä. SCA8:n CTG-jakson pidentymistä todetaan etenkin silloin, kun sairaus on periytynyt äidiltä.
SCA8:n geenimutaatioon liittyy lukuisia kysymyksiä. Joillakin aikuisilla SCA8-perheen jäsenillä, joilla on testissä todettu erittäin pitkä CTG-jakso, ei kliinistä sairautta ole. Tämän havainnon on ajateltu olevan yhteydessä geenimutaation (genotyyppi) heikentyneeseen penetranssiin eli ilmenemiseen (fenotyyppi). On mahdollista, että muutkin perimän tekijät myötävaikuttavat merkittävästi SCA8:n sairausoireiston puhkeamiseen. Sisarussarjassa samanpituisen poikkeavan CTG-jakson omaavat voivat sairastua hyvinkin eri-ikäisinä. Sisarusten oireet eivät etene saman tahtiin.
Antisipaatiolla tarkoitetaan sairauden puhkeamista keskimäärin varhemmin seuraavassa sukupolvessa. Osalla perheistä on havaittu antisipaatiota. Antisipaatio SCA8:ssa on liittynyt pääsääntöisesti äidiltä perittyyn sairauteen. Isältä periytyneen SCA8:n geenimutaation on jopa todettu lyhentyvän seuraavassa sukupolvessa.
DNA-testi varmistaa SCA6:n diagnoosin. DNA-testiä voidaan käyttää myös sairautta ennakoivana (prediktiivinen geenitesti). Määritys voidaan tehdä halutessa täysi-ikäisiltä oireettomilta, riskissä olevilta sukulaisilta tai sikiönäytteestä. Perinnöllisyyslääkärin neuvonta on aina aiheellista ennakoivan geenitestin pohdinnassa.
SCA8:n diagnostiikka ja erotusdiagnostiikka
SCA8-diagnoosiin ja erotusdiagnoosiin tarvitaan neurologinen tutkimus sekä sen perusteella arvioitavat laboratorio-, kuvantamis- ja muut hermotoimintojen tutkimukset. Muiden erikoisalojen konsultaatiot ovat joskus perusteltuja. Perinnöllisyyslääkärin konsultaatio ja neuvonta sairastuneelle ja hänen sukulaisille on aina tarpeen.
Laboratoriotutkimukset verinäytteestä, lukuun ottamatta diagnostista DNA-testiä, ovat normaalit, samoin selkäydinnestenäyte (likvor-tutkimus). Aivojen magneettitutkimuksessa (MRI) rappeutumaa (atrofiaa) nähdään pikkuaivoissa. ENMG-tutkimuksessa todetaan osalla potilaista aksonaalista neuropatiaa.
Erotusdiagnoosissa tärkein ryhmä on muut hitaasti etenevät ja ”puhtaasti” pikkuaivo-oireita aiheuttavat polyglutamiiniataksiat (ADCA III), erityisesti SCA5, SCA6, SCA11, SCA15, SCA26 ja SCA28. SCA8-sairaus todetaan poikkeuksellisen usein myös silloin, kun suvussa ei ataksiasairautta ole aikaisemmastaan todettu. Nämä potilaat edustavat mahdollisesti uusia SCA8-mutaatioita. Toisena vaihtoehtona on, että vanhemman lieväoireinen tai mahdollisesti vasta vanhuusiässä oirehtimaan alkanut sairaus ei ole johtanut diagnoosiin tai geenin penetranssi vanhemmalla on ollut heikko eikä ole aiheuttanut sairautta. SCA8 voi perinnöllisyydeltään muistuttaa myös autosomisesti peittyvästi periytyvää sairautta. Erotusdiagnostisena vaihtoehtona tulevat kyseeseen myös SCA8 sairautta huomattavasti tavallisempi MS-tauti. SCA8 on poikkeuksellisesti todettu potilailla, joiden taudinkuva muistuttaa tyvitumake-sairauksien oireistoa.
SCA8:n esiintyminen
SCA8:aa esiintyy useissa etnisissä ryhmissä ja maailmanlaajuisesti SCA8 esiintyvyyden arvellaan olevan 1 / 100 000, mikä merkitsisi Suomessa 50 sairastavaa. Kaikista SCA-sairauksista SCA8 on osoittautunut Suomessa yleisimmäksi.
SCA8:n hoito ja kuntoutus
Sairauden hoitoon ei ole oireiden etenemistä hillitsevää tai sairautta parantavaa lääkehoitoa. Vapinalääkkeillä ei useinkaan saada lievitystä pikkuaivoperäiseen vapinaoireistoon. Vitamiinilisiä on suositeltu – erityisesti silloin, kun tavanomaisesta ravinnosta niitä ei todennäköisesti riittävästi kerry.
Perinnöllisyysneuvonta on oleellinen osa SCA8:n diagnostiikkaa ja hoitoa. Neuvontaa tarvitsevat sairastuneen lisäksi myös muut perheenjäsenet.
SCA8-sairauden aiheuttamat toimintarajoitteet etenevät yksilöllisesti ja toisaalta sairastumisikä vaihtelee huomattavasti. Tästä syystä kuntoutustoimet on syytä aina arvioida yksilöllisesti samoin kuin tarvittavat palvelut. Pääsääntöisesti SCA8-sairaus etenee hitaasti eikä lyhennä elinikää. Sairaudella voi olla merkitystä ammatillisiin suunnitelmiin sekä työkykyyn.
Puheterapia sekä kommunikaatiota helpottavat apuvälineet voivat tulla ajankohtaiseksi sairauden edetessä. Säännöllisen puheterapian tarve ei useinkaan ole tarpeen, mutta puheterapeutin käynnillä saadut ohjeet voivat merkittävästi selkeyttää ilmaisua. Nielemisvaikeudet kuuluvat useimmilla vasta sairauden myöhäisvuosiin. Puheterapeutti voi antaa ohjeita myös nielemiseen liittyvissä erityiskysymyksissä. Fysioterapeutin neuvonta ja säännöllinen liikunta tukevat liikuntakyvyn säilymistä. Sairauden edetessä liikunnan apuvälinetarve on arvioitava.
SCA8-ataksiasairaudessa on todettu muistiin sekä laajemminkin kognitiiviseen suoriutumiseen liittyviä muutoksia. Epäillyt muutokset ja niiden laatu ja vaikeusaste on syytä varmentaa neuropsykologisella tutkimuksella. Käyttäytymiseen ja mielialaan liittyviä häiriöitä voidaan hoitaa oireenmukaisilla lääkkeillä.
SCA8:n perustutkimus
SCA8:n neuropatologisia tutkimusraportteja ei toistaiseksi ole käytettävissä. Aivojen magneettitutkimuksessa muutokset paikallistuvat ennen kaikkea pikkuaivoihin säästäen muut aivoalueet.
SCA8 geeni sijaitsee kromosomissa 13 alueella q21. SCA8:n geenimutaatio – pidentynyt CTG-jakso – dokumentoitiin alun perin vuonna 1999 joissakin ataksiaperheissä, joiden perinnöllisen ataksian syy ei ollut selvinnyt aikaisemmin tunnettuja SCA-geenimutaatioita kartoitettaessa. Normaaleissa geenin vastinpareissa CTG-jaksojen pituus vaihtelee 16–34 CTG-jaksoon. Sairastuneilla CTG-jakson pituus on yleensä 100 tai enemmän. SCA8-geeni ohjaa aktiiniin sitoutuvan valkuaisaineen Kelch-like 1-proteiinin (KLHL1) tuotantoa. KLHL1 proteiinia esiintyy erityisesti hermosoluissa myös pikkuaivoissa. KLHL1-proteiini paikallistuu soluissa solulimaan ja sillä ajatellaan olevan merkitystä solujen aktiini-sytoskeletoni järjestyksen ylläpitäjänä.
Useissa tutkimuksissa on yksittäisillä kontrolliaineistoon kuuluvilla henkilöillä todettu niin ikään pidentynyt CTG-jakso. Pidentynyt CTG-jakso on todettu myös henkilöillä, joilla on alun perin diagnostisoitu muu neurologinen sairaus (perinnöllinen polyneuropatia, Friedreichin ataksia, E-vitamiinin puutoksesta aiheutuva ataksia, sporadinen etenevä ataksiasairaus jne.). SCA8:n geenimutaatioon liittyy edelleen useita avoimia kysymyksiä.
Kirjoittaja
Riitta Rinne, LL, neurologian erikoislääkäri, Maskun neurologinen kuntoutuskeskus
Julkaistu: 20.11.2002. Päivitetty: 16.6.2006
- Kirjallisuusviitteet
- Koob MD et al., An untranslated CTG expansion causes a novel form of spinocerebellar ataxia
(SCA8). Nature genetics 21: 379–384, 1999 - Day JW et al., Spinocerebellar ataxia type 8. Clinical features in a large family. Neurology 55: 649–657, 2000
- Ikeda Y et al., Molecular and clinical analyses of spinocerebellar ataxia type 8 in Japan. Neurology
54: 950–955, 2000 - Juvonen V et al., Clinical ans Genetic Findings in Finnish Ataxia Patients with the Spinocerebellar
Ataxia 8 Repeat Expansion. Ann Neurol 48: 354–361, 2000 - Moseley ML et al., SCA8 CTG repeat: en masse contractions in sperm and intergenerational
sequence changes may play a role in rduced penetrance. Hum Mol Genet 9: 2125–2130, 2000 - Nemes JP et al., The SCA8 transcript is an antisense RNA to a brain‐specific transcript encoding a
novel actin‐binding protein (KLHL1). Hum Mol Genet 9: 1543–1551, 2000 - Silveira I et al., High germinal instability of the (CTG)n at the SCA8 locus of both expanded and
normal alleles. Am J Hum Genet 66: 830–840, 2000 - Stevanin G et al., Are (CTG)n expansions at the SCA8 locus rare polymorphisms? Nature Genetics
24: 213, 2000 - Vincent JB et al., An unstable trinucleotide‐repeat region on chromosome 13 implicated in
spinocerebellar ataxia: a common expansion locus. Am J Hum Genet 66: 819–829, 2000 - Worth PF et al., Large, expanded repeats in SCA8 are not confined to patients with cerebellar
ataxia. Nature Genetics 24: 214–215, 2000 - Cellini A et al., Genetic and Clinical Analysis of Spinocerebellar Ataxia Type 8 Repeat Expansion in
Italy. Arch Neurol 58 : 1856–1859, 2001 - Sorbido M‐J et al., SCA 8 repeat expansions in ataxia: A controversial association. Neurology 57:
1310–1312, 2001 - Tan E‐K and Ashizawa T: Genetic testing in spinocerebellar ataxias. Defining a clinical role. Arch
Neurol 58: 191–195, 2001 - Cellini A et al., A Family with spinocerebellar ataxia type 8 expansion and vitamin E deficiency
ataxia. Arch Neurol 59: 1952–1953, 2002 - Izumi Y et al., SCA8 repeat expansion: Large CTA/CTG repeat alleles are more common in ataxic
patients, including those with SCA6. Am J Hum Genet 72: 704–709, 2003 - Mosemiller AK et al., Molecular genetics of spinocerebellar ataxia type 8 (SCA8). Cytogenet
Genome Res 100: 175–183, 2003 - Schols L et al., Do CTG expansions at the SCA8 locus cause ataxia? Ann Neurol 54: 5–7, 2003
- Wu YR et al., Genetic testing in spinocerebellar ataxia in Taiwan: expansion of trinucleotide
repeats in SCA8 and SCA17 are associated with typical Parkinson´s disease. Clin Genet 65: 209–214, 2004 - Zeman A et al., Spinocerebellar ataxia type 8 in Scotland: genetic and clinical features in seven
unrelated cases and a review of published repots. J Neurol Neurosurg Psychiatry 73: 459–465,
2004 - Baba Y et al., Sporadic SCA8 mutation resembling corticobasal degeneration. Parkinson Relat
Disord 11: 147–150, 2005 - Juvonen V et al. The occurence of dominant spinocerebellar ataxias among 251 Finnish ataxia
patients and the role of predisponding large normal alleles in a genetically isolated population.
Acta Neurol Scand 111: 154–1562, 2005 - Lilja A et al., Cognitive impairment in spinocerebellar ataxia type 8. J Neurol Sci 237: 31–38, 2005
- Koob MD et al., An untranslated CTG expansion causes a novel form of spinocerebellar ataxia
Järjestötoiminta
Potilasjärjestö: Neuroliitto ry; ataksiaverkosto
Muita tietolähteitä
- The National Ataxia Foundation: www.ataxia.org
- Sveriges socialstyrelsens kunskapsdatabas om ovanliga diagnoser innehåller information om fler än 300 ovanliga sjukdomar och tillstånd.