
Spinoserebellaarinen ataksia tyyppi 7 (SCA7)
- ICD-10: G11.8 Muut määritetyt perinnölliset ataksiat, OMIM: 164500, ORPHA: 94147
- Muut sairaudesta käytetyt nimet:
- Olivopontoserebellaarinen atrofia III; OPCA3
- OPCA III
- OPCA ja retinaalinen degeneraatio
- OPCA ja makuladegeneraatio sekä eksterni oftalmoplegia
- Autosomaalinen dominantti serebellaarinen ataksia, tyyppi II
- ADCA, tyyppi II
Spinoserebellaarinen ataksia tyyppi 7 (SCA7) on pikkuaivojen ja niiden hermoratayhteyksien toimintahäiriöihin johtava etenevä neurologinen sairaus, jonka oirekuvaan liittyy aina verkkokalvon rappeuma ja näön heikkeneminen (ADCA II).
Sairaus periytyy autosomisesti vallitsevasti eli dominantisti (AD). Sairauden oireiden alkaminen ja niiden eteneminen vaihtelevat sairastuneilla merkittävästi. Näön heikkeneminen voi alkaa jo ennen tasapainovaikeutta. Pikkuaivo-oireiden (tasapainovaikeudet, hienomotoriikan kömpelyys ja puheen epäselvyys) lisäksi silmien liikehäiriöt, raajojen lisääntynyt tonus (spastisiteetti), tahattomat liikkeet sekä syvätuntojen heikkeneminen kuuluvat oirekuvaan. Sairautta aiheuttava geenimutaatio tunnetaan ja sen määrittäminen verinäytteestä varmistaa SCA7-diagnoosin. Geenimutaation perusteella SCA7 lukeutuu polyglutamiiniataksioihin (PolyQ-ataksiat). SCA7 on Suomessa toiseksi tavallisin aikuisiän AD-periytyvä etenevä ataksiasairaus.
SCA7 alkaa tavallisimmin keski-ikäisenä (30–40 vuoden iässä), mutta voi periaatteessa tapahtua missä iässä tahansa (3 kk–60 v). Mikäli SCA7 alkaa jo lapsena, kliiniset oireet eivät ilmene samankaltaisina kuin aikuisiällä puhkeavassa oireistossa. Aikuisiällä ensioireena todetaan tavallisimmin tasapainovaikeudet, hienomotoriikan heikkous ja vähittäisesti etenevät näön ongelmat. Lapsuudessa tai nuoruusvuosina sairaus alkaa näön heikkenemisellä ja tasapainovaikeudet kehittyvät vasta myöhemmin. Sairauden puhkeaminen lapsuudessa ennen puberteettia johtaa näön heikkenemiseen ja sokeutumiseen mahdollisesti jo muutaman vuoden kuluessa.
Liikuntaongelmat aiheuttavat keskimäärin 10 vuoden kuluessa apuvälinetarvetta. SCA7 lyhentää useimpien kohdalla elinikää ja oireiden kesto on keskimäärin 20 vuotta (1–45 vuotta). Samassakin perheessä sisarusten sairastumisikä vaihtelee huomattavasti, samoin kuin oireiden eteneminen ja niiden moninaisuus (Kuva 1.). Sairastumisiän ja oireiden etenemisnopeuden välillä on todettu selvää korrelaatiota: mitä varhemmin sairaus puhkeaa, sitä todennäköisempää on sen nopea eteneminen.
Aikuisiän SCA7:n taudinkuvaan kuuluvat
- tasapainon ja kävelyn epävarmuus (kävelyataksia)
- vartalon ja raajojen liikkeiden hallintavaikeus (ataksia)
- osumatarkkuuden heikkeneminen (dysmetria) ja vuorottaisten liikesuoritusten epärytmisyys (dysdiadokokinesis)
- hienomotoriikan kömpelyys ja hitaus
- puheen kangertaminen ja puherytmin hitaus(dysartria)
- alaraajojen lievä lihasjänteyden nousu (spastisuus) sekä heijasteiden kiihtyminen (hyperrefleksia)
- verkkokalvon rappeutuma, muutoksia värinäössä, terävän näön keskuksen rappeutumista (makuladegeneraatio)
- kognitiivista heikentymistä
- nielemisen ongelmat (dysphagia)
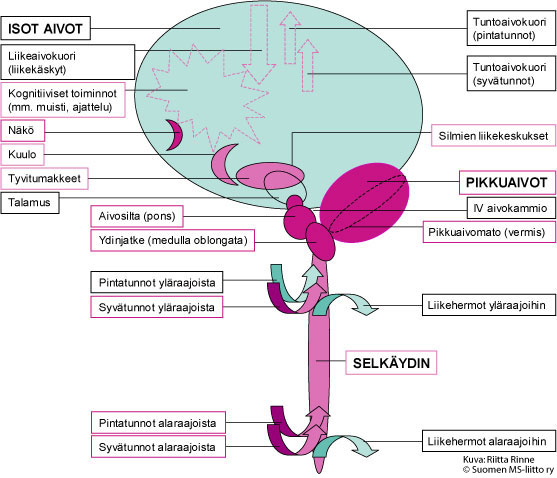
Kuva 1. Aikuisiällä alkavan SCA7:n aiheuttama neurologinen oireisto johtuu pikkuaivojen ja niiden hermoratayhteyksien toiminnan heikkenemisestä sekä verkkokalvon rappeutumisesta. Kuvassa on tummalla violetilla merkitty hermoston alueet, joissa SCA7-sairaus aiheuttaa aina muutoksia ja vaaleammalla violetilla ne hermoalueet, joissa muutoksia voi esiintyä.
SCA7:n taudinkuva ja oireiden eteneminen
SCA7 diagnoosia voidaan pitää kohtuullisen varmana kliinisen tutkimuksen perusteella. Moni aikuisiällä sairastunut on mahdollisesti vuosia ennen tutkimuksiin hakeutumista todennut ongelmia juoksemisessa, pallopeleissä tai vaativissa hienomotorisissa suorituksissa (esim. koneella kirjoittaminen). Hienoiset tasapainon ongelmat havaitaan kompurointina maastossa kävellessä, portaita laskeutuessa. Äkillisissä käännöksissä horjahtaa herkästi ja tarvitaan tukiaskeleita. Kävellessä jalat voivat tuntua jäykiltä ja askeltaminen hidastuu ja muuttuu leveäraiteiseksi. Vartalonhallinnassa on ongelmia (vartaloataksia). Vuosien mittaan tarvitaan liikunnan apuvälineitä. Ensioireisiin ja eteneviin oireisiin kuuluvat käsien kömpelyys. Hienomotoriikka hidastuu. Uusien liiketaitojen oppiminen ei onnistu.
Puherytmi hidastuu, artikulointi muuttuu kangertavaksi, hitaaksi ja sammaltavaksi. Puheen kangerrus todetaan yleensä jo sairastumisen ensi vuosien aikana. Puheen sisältö ei muutu eikä puheen ymmärtämisessä ilmene vaikeutta. Nielemisvaikeuksia todetaan sairauden edetessä. Ne aiheuttavat ruuan ja erityisesti nesteiden nielaisuna ”väärään kurkkuun”. Sairauteen liittyy painon putoamista, joka ei välttämättä ole yhteydessä nielemisen ongelmiin.
Silmienliikkeet rajoittuvat ja katseen kohdistaminen vaikeutuu. Neuro-oftalmologisessa tutkimuksessa verkkokalvojen rappeutumaa todetaan jo ennen näön heikkenemistä. Muutokset kehittyvät pikkuhiljaa. Ensimmäiseksi havaitaan ongelmia värinäössä ja jatkossa näkökyky vähittäisesti menetetään kokonaan. Näön aleneminen johtuu terävän näön keskusten (makulan) ja verkkokalvojen rappeutumisesta. Näköoireiden etenemisnopeus on yhteydessä sairauden varhaiseen puhkeamiseen: näkökyky heikkenee suhteellisesti nopeammin varhain alkaneessa sairaudessa. Tunnetaan lapsena sairastuneita, jotka ovat sokeutuneet muutaman vuoden kuluessa. Sairastuneiden vastasyntyneiden (alle 1 vuoden ikäisten) sairauden muiden oireiden eteneminen saattaa tapahtua niin nopeasti, ettei näköä kyetä luotettavasti tutkimaan. Puberteetti-iän ohittaneen näön heikkeneminen tapahtuu suhteellisesti hitaammin. Näön heikkeneminen on hitainta keski-ikäisenä sairastuneilla.
Kuulon alentumista, jopa kuuroutumista on todettu satunnaisesti SCA7:ssä. Käytännössä kuulon aleneminen aiheuttaa ongelmia puheen ymmärtämisessä etenkin meluisassa ympäristössä. Joillakin SCA7:ää sairastavilla on todettu ongelmia kognitiivisessa suoriutumisessa, mutta mahdollisia sairauteen liittyviä kognitiivisia ongelmia ei ole systemaattisesti kartoitettu.
Autonomisen hermoston toimintahäiriöitä on kartoitettu niukasti. Verenpaineen säätelyyn, hikoiluun sekä rakontoimintaan liittyviä häiriöitä on todettu SCA 7:ssä. Ortostaattista hypotoniaa eli verenpaineen romahtaminen makuulta seisomaan noustessa on todettu osalla potilaista. Seurauksena on huimausta tai hankalimmillaan tajunnan menetykseen johtava verenpaineen lasku. Rakontoimintahäiriöt voivan niin ikään liittyä oirekuvaan.
Alle on koottu SCA7:ään liittyviä kliinisiä oireita
SCA7:n kliiniset oireet 71 sairastavalla, 20 eri SCA7-perheestä. Tutkittujen ikä oli 38,9 ± 18,4 v. (2–85 v.). Pikkuaivo-oireita oli 66:lla ja niiden alkamisikä oli 30,3 ± 15,9 v. (1–70 v.). Näön heikkeneminen todettiin 47:llä ja sen alkaminen oli 28,9 ± 17,5 v. (1–69 v.). Lähde: David G et al., 1998.
- Kävelyataksia 100 %
- Raajojen ataksia 95 %
- Puheen sammaltaminen 98 %
- Näön heikkeneminen 83 %
- Silmien seurantaliikkeiden hitaus 88 %
- Alaraajojen vilkastuneet heijasteet 78 %
- Heikentynyt värinätunto 62 %
- Nielemisvaikeuksia 58 %
- Silmien liikerajoituksia 53 %
- Ojentava jalkapohjaheijaste 52 %
- Rakon toimintahäiriöitä 50 %
- Alaraajojen lihastonuksen nousu 41 %
- Alaraajojen lihaskato 25 %
- Kuulon aleneminen 24 %
- Posturaalinen vapina 19 %
- Parkinsonin taudin kaltaisia oireita 18 %
- Kasvojen lihasnykäyksiä (myokymiaa) 16 %
- Kognitiivisia muutoksia 11 %
- Selkärangan kiertymistä (skolioosi) 11 %
Nuorimmat SCA7 diagnoosin saaneet ovat alle vuoden ikäisiä. Imeväisiällä tai varhaislapsuudessa alkanut sairaus johtaa usein nopeaan liikunnallisten aktiviteettien taantumiseen sekä näön heikkenemiseen ja sokeutumiseen. Vastasyntyneisyyskaudella alkaneeseen SCA7 sairauteen liittyvät lihasvelttous, imemisvaikeudet ja hengityksen ongelmat sekä sydänvika. Vastasyntyneen SCA7 on nopeasti kuolemaan johtava tila. Lapsena alkaneeseen sairauteen liittyvät myös epileptiset kohtaukset, myoklonia eli lihasnykäykset sekä henkinen taantuminen. Painon putoamista on todettu.
SCA7:n periytyminen
SCA7 periytyy autosomisesti vallitsevasti eli dominantisti (AD). Sairautta aiheuttava geenivirhe tunnistettiin kromosomissa 3(p21.1 – p12) vuonna 1997. Geenitesti varmistaa SCA7:n diagnoosin. SCA7-sairauden geeni on ATXN7 ja sen tuottama valkuaisaine on Ataksiini – 7. SCA7-sairaudessa geenimutaatio on geenin luentakehyksen sisäinen ylipitkä, epävakaa CAG-jakso (dynaaminen mutaatio). Normaalissa ATXN7-geenissä on alle 20 CAG-jaksoa (tavallisimmin 10 CAG-jaksoa). SCA7-sairauden geenissä on yli 35 CAG-jaksoa. Tavallisimmin sairastuminen todetaan vasta, jos CAG-jaksoja on > 38. CAG-nukleotidikomikko ohjaa aminohappo glutamiinin syntymisen. Seurauksena on ylipitkä glutamiiniketju (PolyQ-ketju) Ataksiini -7:ssä (Kuva2.).
Ylipitkän CAG-jakson dynaamisuus merkitsee CAG-jakson taipumusta pidentyä seuraavassa sukupolvessa. CAG-jakson pituuden ja oireiden puhkeamisen välillä on todettu käänteistä korrelaatiota: pitkä CAG-jakso on yhteydessä oireiden varhaiseen puhkeamiseen ja nopeaan taudinkulkuun (genotyyppi – fenotyyppi korrelaatio). Sairauden varhaistumista seuraavassa sukupolvessa kutsutaan antisipaatioksi. CAG-jakson pidentymistä on havaittu etenkin sairauden periytyessä isältä.
CAG-jakson pituuden perusteella ei oireiden puhkeamisikää tai niiden etenemistä voi täysin ennustaa. Kaikkein pisimmät CAG-jaksot (yli 200 jaksoa) on todettu vastasyntyneen SCA7-sairaudessa. Suurta vaihtelua niin oireiden ilmaantumisessa kuin myös etenemisessä on havaittu sisaruksilla, joiden CAG-jakson pituus on sama. CAG-jakson pituus ei siis yksinomaan selvitä SCA7 sairauden oirekulkua. 30 – 36 pituinen CAG-jakso edustaa geenin harmaata aluetta. Näillä henkilöillä voidaan satunnaisesti todeta lieviä oireita ja löydöksiä, jotka sopivat SCA7 sairauteen. (CAG)30 – 36 voi olla geenin esimutaatio ja jakson pidentyminen seuraavassa sukupolvessa on mahdollista. Harmaan alueen omaavien mahdollisia oireita tunnetaan kovin huonosti. SCA7:ssä on todettu uusien mutaatioiden osuudeksi peräti 10%.
DNA-testi varmistaa SCA7:n diagnoosin. DNA-testiä voidaan käyttää myös sairautta ennakoivana (ennustava eli prediktiivinen geenitesti). Määritys voidaan tehdä halutessa täysi-ikäisiltä oireettomilta, riskissä olevilta sukulaisilta tai sikiönäytteestä.
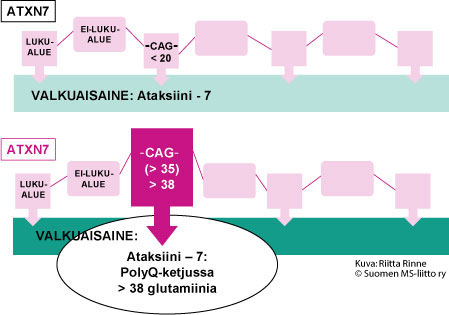
Kuva 2. SCA7-sairaudessa geenivirhe aiheuttaa Ataksiini-7 valkuaisaineeseen ylipitkän PolyQ-ketjun syntymisen.
SCA7:n diagnoosi ja erotusdiagnoosi
SCA7:n diagnoosiin tarvitaan neurologinen ja neuro-oftalmologinen tutkimus sekä niiden perusteella arvioitavat laboratorio-, kuvantamis- ja muut hermotoimintojen tutkimukset. Muiden erikoisalojen konsultaatiot ovat usein perusteltuja, mutta periaatteessa SCA7-sairauden kliininen diagnoosi selviää ilman niitä. Perinnöllisyyslääkärin konsultaatio ja neuvonta sairastuneelle ja sukulaisille on aina tarpeellista..
Laboratoriotutkimukset verinäytteestä, lukuun ottamatta diagnostista DNA-testiä, ovat normaalit, samoin selkäydinnestenäyte (likvor-tutkimus). Magneettitutkimuksessa todetaan rappeutumaa pikkuaivojen ja aivorungon alueella (olivopontoserebellaarinen atrofia). Neuro-oftalmologin tutkimuksessa todetaan ensivaiheessa verkkokalvon rappeuma ja vähäiset jyvämäiset muutokset makulassa. Ongelmia on värinäössä (sininen – keltainen -erotus heikkenee). Elektroretinogrammi-tutkimuksessa muutokset todetaan yleensä varhemmin tappien kuin sauvojen antamassa vasteessa. Neurofysiologisissa tutkimuksissa todetaan joko tuntohermoihin tai sekä tunto- että liikehermoihin painottuva aksonaalinen polyneuropatia.
Erotusdiagnoosissa muista SCA-sairauksista tulee kyseeseen lähinnä SCA1, jossa varsinkin nuorilla sairastuneilla on todettu näön heikkenemistä. Mitokondriaalinen Leberin tauti on huomioitava erotusdiagnostiikassa. Lapsena alkavan SCA7-sairauden erotusdiagnoosiin kuuluvat mm. neuronaaliset lipofuskinoosit.
SCA7:n esiintyminen
Suomessa SCA7 on toiseksi yleisin SCA-sairauksista (12,5 %). Myös Ruotsissa SCA7-diagnoosia on tehty keskimääräistä enemmän. Sekä suomalaiset että ruotsalaiset SCA7- perheet polveutuvat samasta kantaisästä. Muualla Euroopassa samoin kuin Amerikassa, Kanadassa, Australiassa ja Kiinassa SCA7 on erittäin harvinainen. Etelä-Afrikassa SCA7 osuus on peräti 22 % SCA-sairauksista, Koreassa vastaavat esiintyvyysluvut ovat 11–16 % ja Meksikossa 11 %.
SCA7:n aiheuttamat hermovauriot
Neuropatologisiset muutokset todetaan pikkuaivojen, aivorungon, talamuksen, tyvitumakkeiden sekä selkäytimen alueella ja myös paikoittain aivokortexilla. Alimpien oliva-tumakkeiden solutuho on huomattava. Spinoserebellaariset, olivoserebellaariset ja efferentit pikkuaivoradat ovat rappeutuneet. Pontoserebellaarisissa ratayhteyksissä ei rappeutumista todeta. Pikkuaivoissa rappeumaa todetaan merkittävämmin pikkuaivomadossa (vermiksessa) kuin pikkuaivokupoleissa (hemisfääreissä). Pikkuaivoissa todetaan niin Purkinjen solujen kuin vähäisemmässä määrin myös granule-solujen tuhoa. Verkkokalvossa ja näköhermossa nähdään solutuhoa.
SCA7:n hoito ja kuntoutus
Ataksian lievittäminen käytössä olevin lääkkein ei onnistu. Vapinaoireeseen voi yrittää beeta-salpaajaa. Parkinsonin taudin lääkehoidossa käytettävät L-dopamiini tai dopamiinin vaikutusta tehostavat valmisteet (dopamiini-agonistit) voivat lievittää SCA7-sairauteen liittyviä Parkinsonin sairauden kaltaisia oireita ja jalkojen levottomuutta. Rakon toimintahäiriöihin on niin ikään saatavissa apua. Aloitettavan oirelääkkeen tehon arvioinnissa on oltava kriittinen. Etenevässä sairaudessa lääkkeen tarpeellisuutta ja sen annosta on syytä tarkistaa. Hoitava lääkäri antaa ohjeet annosmäärän säätelyyn. Pitkään käytössä olleiden keskushermostoon vaikuttavien lääkkeiden lopettamista ei saa tehdä äkillisesti.
Kuntoutustarpeen arvioiminen ja kirjaaminen kuntoutussuunnitelmaan ja seuranta ovat keskeinen osa SCA7-sairauden hyvää hoitoa. Kuntoutussuunnitelmassa on arvioitava eri terapioiden tarve, tarvittavat apuvälineet ja muut selviytymistä tukevat keinot. Tarvittavat palvelut kirjataan palvelusuunnitelmaan.
SCA7:ää sairastava tarvitsee opastusta näönkäyttöön sekä arvion ympäristön valaistuksen optimoinnista. Näön heikkeneminen on etenevää, joten säännölliset näkökontrollit ovat välttämättömiä sairastuneen turvallisen liikkumisen, samoin kuin muunkin turvallisuuden takia. Liikuntakyvyn säilymistä tuetaan fysioterapialla. Tasapainon, lihasten hallinnan sekä koordinaation harjoittaminen omaehtoisesti kannattaa. Soveltuvista liikunnan muodoista saa ohjeita fysioterapeutilta, samoin kuin opastusta lihashuollosta ja rentoutuksesta. Puheterapeutin arviot ja ohjeet ylläpitävät kommunikaatiotaitoja ja keinoja on löydettävissä myös turvalliseen nielemiseen. Nielemisvaikeuksien takia ruoka voi mennä ”väärään kurkkuun” eli henkitorveen (aspiraatio). Aspiraatio altistaa toistuville keuhkotulehduksille ja pahimmillaan tukehtumiselle. Aspiraation riski tutkitaan videofluorometrialla. Nielemisen turvallisuuteen voidaan vaikuttaa. Nielemisen koordinointiongelmissa on vältettävä karkeusasteeltaan vaihtelevia ruokia. Kiinteä ruoka ja nesteet on nautittava erikseen. Nielemisen turvallisuutta lisäävät ruuan ja nesteiden tasalaatuisuus ja ruokailun rauhoittaminen. Vatsanpeitteiden läpi asetettavaa PEG-letkua suositellaan silloin, jos ruokailu aspiraatiovaaran takia ei ole turvallista.
Niin kommunikaatiota, näköä, arkipäivän toimien suorittamista kuin myös liikkumista on mahdollista helpottaa apuvälinein. Sairauden aiheuttaessa lisääntyvää avun- ja hoivan tarvetta on tehtävä palvelusuunnitelma. Kuntoutussuunnitelman tavoin myös palvelusuunnitelman ajantasaisuus on sovitusti tarkistettava.
SCA7:n perustutkimuksesta
SCA7-sairauden perustutkimukseen on kehitetty niin hiiri- kuin kärpäsmalleja. SCA7-geeniä todetaan käytännöllisesti kaikissa kudoksissa. Runsaasti SCA7-geeniä on sydämessä, istukassa, lihaskudoksessa sekä haimassa. SCA7-geeniä on myös aivoissa, maksassa ja munuaisissa ja verkkokalvossa. Keskushermostossa sitä todetaan erityisesti pikkuaivoissa. Ataksiini-7 paikallistuu aivokudoksessa hermosoluihin. Virheellinen ataksiini-7 saostuu solulimaan ja saostumia todetaan myös tumissa. Tumansisäisillä Ataksiini-7 saostumilla ajatellaan olevan suoranaista merkitystä sairauden kehittymiselle, mutta virheellinen Ataksiini-7 voi muutoinkin aiheuttaa solutoiminnalle haittaa.
Kirjoittaja
Riitta Rinne, LL, neurologian erikoislääkäri, Maskun neurologinen kuntoutuskeskus
Julkaistu: 1.11.2002, Päivitetty: 16.6.2006
- Kirjallisuusviitteet
- Benomar A et al., Autosomal‐dominant cerebellar ataxia with retinal degeneration (ADCA type II)
is genetically different from ADCA type I. Ann Neurol 35: 439–444, 1994 - Evevoldson TP et al., Autosomal dominant cerebellar ataxia with pigmentary macular dystrophy. A
clinical and genetic study of eight families. Brain 117: 445–460, 1994 - Benomar A et al., The gene for autosomal dominant cerebellar ataxia with pigmentary macular
dystrophy maps to chromosome 3p12‐p21.1. Nat Genet 10: 84–88, 1995 - Gouw LG et al., Retinal degeneration characterizes a spinocerebellar ataxia mapping to
chromosome 3p. Nat Genet 10: 89–93, 1995 - David G et al., The gene for autosomal dominant cerebellar ataxia type II is located in a 5cM region
in 3p12 – p13: genetic and physical mapping of the SCA7 locus. Am J Hum Genet 59: 1328–1336,
1996 - Lindblad K et al., An expanded CAG repeat sequence in spinocerebellar ataxia type 7. Genome Res
6: 965–971, 1996 - David G et al., Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat
Genet 17: 65–70, 1997 - Kaakkola S ja Rinne R: Ataksiat ja niiden erotusdiagnostiikka (katsaus). Duodecim 113: 1773–1782, 1997
- Benton CS et al., Molecular and clinical studies in SCA‐7 define a broad clinical spectrum and the
infantile phenotype. Neurology 51: 1081–1086, 1998
David G et al., Molecular and clinical correlations in autosomal dominant cerebellar ataxia with
progressive macular dystrophy (SCA7). Hum Mol Genet 7: 165–170, 1998 - Gouw LG et al., Analysis of the dynamic mutation in the SCA7 gene shows marked parental effects
on CAG repeat transmission. Hum Mol Genet 7: 525–532, 1998 - Johansson J et al., Expanded CAG repeat in Swedish spinocerebellar ataxia type 7 (SCA7) patients:
effect of CAG repeat length on the clinical manifestation. Hum Mol Genet 7: 171–176, 1998 - Holmberg M et al., Spinocerebellar ataxia type 7 (SCA7): a neurodegenerative disorder with
neuronal intranuclear inclusions. Hum Mol Genet 7: 913–918, 1998 - Klockgether T et al., The natural history of degenerative ataxia: a retrospective study in 466
patients. Brain 121: 589–600, 1998 - Moseley ML et al., Incidence of dominant spinocerebellar and Friedreich triplet repeats among
361 ataxia families. Neurology 51: 1666–1671, 1998 - Jonasson J et al., Evidence for common spinocerebellar ataxia type 7 (SCA7) founder mutation in
Scandinavia. Eur J Hum Genet 8: 918–922, 2000 - Modi G et al., The clinical and genetic characteristics of spinocerebellar ataxia type 7 (SCA7) in
three Black South African Families. Acta Neurol Scand 101: 177–182, 2000 - Potter NT and Nance MA: Genetic testing for ataxia in North America. Mol Diagn 5: 91–99, 2000
- Stevanin G et al., Spinocerebellar Ataxia Type 7. Kirjassa: Handbook of Ataxia Disorders, 470–486.
Ed. Klockgether T, Marcel Dekker Inc., New York‐Basel, 2000 - Storey E et al., Frequency of spinocerebellar ataxia types 1, 2, 3, 6, and 7 in Australian patients
with spinocerebellar ataxia. Am J Med Genet 95: 351–357, 2000 - Tang B et al., Frequency of SCA1, SCA2, SCA3/MJD, SCA6, SCA7, and DRPLA CAG trinucleotide
repeat expansions in patients with hereditary spinocerebellar ataxia from Chinese kindreds. Arch
Neurol 57: 540–544, 2000 - Jardim LB et al., A survey of spinocerebellar ataxia in South Brazil – 66 new cases with Machado‐
Joseph disease, SCA7, SCA8, or unidentified disease‐causing mutations. J Neurol 248: 870–876,
2001 - Oh AK et al., Slowing of voluntary and involuntary saccades: an early sign in spinocerebellar ataxia
type 7. Ann Neurol 49: 801–804, 2001 - Yvert G et al., SCA7 mouse models show selective stabilization of mutant ataxin‐7 and similar
cellular responses in different neuronal cell types. Hum Mol Genet 10: 1679–1692, 2001 - Bang OY et al., Pontine atrophy precedes cerebellar degeneration in spinocerebellar ataxia 7: MRI‐
based volumetric analysis. J Neurol Neurosurg Psychiatry 75: 1452–1456, 2004 - Helmlinger D et al., Disease progression despite early loss of polyglutamine protein expression in
SCA7 mouse model. J Neurosci 24: 1881–1887, 2004 - Helmlinger D et al., Hsp70 and Hsp40 Chaperones do not modulate retinal phenotype in SCA7
mice. J Biol Chem 279: 55969–55977, 2004 - Bowman AB et al., Neuronal dysfunction in a polyglutamine disease model occurs in the absence
of ubiquitin‐proteasome system impairment and inversely correlates with the degree of nuclear
inclusion formation. Hum Mol Genet 14: 679–691, 2005 - van de Warrenburg BPC et al., Peripheral nerve involvement in spinocerebellar ataxias. Arch
Neurol 61: 257–261, 2004 - Juvonen V et al. The occurence of dominant spinocerebellar ataxias among 251 Finnish ataxia
patients and the role of predisponding large normal alleles in a genetically isolated population.
Acta Neurol Scand 111: 154–1562, 2005 - Mascke M et al., Clinical feature profile of spinocerebellar ataxia type 1 – 8 predicts genetically
defined subtypes. Mov Disord 20: 1405–1412, 2005 - Rub U et al., Spinocerebellar ataxia type 7 (SCA7): first report of a systematic neuropathological
study of the brain of a patient with a very short expanded CAG‐repeat. Brain Pathol 15: 287–295,
2005 - van de Warrenburg BPC et al., Age at onset variance analysis in spinoserebellar ataxias: a study in
a Dutch‐French cohort. Ann Neurol 57: 505–512, 2005
Crum BA and Josephs KA: Varied electrophysiologic patterns in spinocerebellar ataxia type 2. Eur J
Neurol 13: 104–197, 2006
- Benomar A et al., Autosomal‐dominant cerebellar ataxia with retinal degeneration (ADCA type II)
Järjestötoiminta
Potilasjärjestö: Neuroliitto ry; ataksiaverkosto
Muita tietolähteitä
- The National Ataxia Foundation: www.ataxia.org
- Sveriges socialstyrelsens kunskapsdatabas om ovanliga diagnoser innehåller information om fler än 300 ovanliga sjukdomar och tillstånd.