
Spinoserebellaarinen ataksia tyyppi 6 (SCA6)
- ICD-10: G11.8 Muut määritetyt perinnölliset ataksiat, OMIM: 183086, ORPHA: 98758
Spinoserebellaarinen ataksia tyyppi 6 (SCA6) on pikkuaivojen toimintahäiriöstä johtuva etenevä neurologinen sairaus (ADCA III). SCA6 periytyy autosomisesti vallitsevasti eli dominantisti (AD). Pääsääntöisesti SCA6:n oireet rajoittuvat tasapaino- ja kävelyvaikeuksiin, puheen sammaltamiseen ja silmänvärveeseen (nystagmukseen). Sairaus etenee hitaasti eikä lyhennä elinikää. Sairautta aiheuttava geenimutaatio tunnetaan ja sen määrittäminen verinäytteestä varmistaa diagnoosin. SCA6 lukeutuu polyglutamiiniataksioihin (PolyQ-ataksiat). SCA6:n geenimutaatio aiheuttaa toimintahäiriön kalsium-kanavassa, mistä syystä SCA6 on myös ionikanavasairaus.
SCA6 alkaa tyypillisesti vasta 40. tai vasta 50. ikävuoden jälkeen, keskimäärin 52 ± 2 vuoden ikäisenä. Oireiden alkamisiässä on suurta vaihtelua (19–71 v). Jokseenkin poikkeuksetta SCA6 on hidaskulkuinen sairaus eikä vaikuta sairastavan elinikään. Miesten ja naisten oireiden alkamisiässä ei ole eroa. Liikunnan apuvälineet tulevat tarpeellisiksi runsaan kymmenen sairastamisvuoden jälkeen (km. 14–15 v), mutta sairastamisaika ilman apuvälineitä voi olla huomattavasti pidempi. Pitkään jatkuneeseen sairauteen voi liittyä myös nielemisongelmia ja silmien liikerajoituksia.
Aikuisiän SCA6:n taudinkuvaan kuuluvat
- tasapainon ja kävelyn epävarmuus (kävelyataksia)
- vartalon ja raajojen liikkeiden hallintavaikeus (ataksia)
- puheen sammaltaminen ja artikulaation epäselvyys (dysartria)
- rytminen silmänvärve (nystagmus) sivuille katsottaessa
Lisäksi voidaan todeta:
- tahattomia liikehäiriöitä ja pakkoliikkeitä (dystoniaa)
- refleksimuutoksia
- tunnon alenemaa
- katseen kohdistamisen vaikeutta
SCA6:n taudinkuva ja oireiden eteneminen
Lähes kaikilla SCA6-potilailla todetaan ensioireena tasapainon epävarmuus ja liikkumisen vaikeudet (Kuva 1.). Vartaloataksia on nystagmuksen ohella SCA6:ssa tyyppilöydös. Sairastamisen ensivuosina koordinaatiohäiriöt voivat olla varsin vähäisiä ja todettavissa kaikkien raajojen yhtäaikaisissa suorituksissa (juokseminen, hiihtäminen, tanssiminen jne.). Hienoinen tasapainohaitta havaitaan rappuja laskeutuessa ja horjahteluna käännöksissä.
Käsien osumatarkkuus heikkenee ja tekeminen vaatii keskittymistä. Käsien kohdistusvapina (intention tremor) on tavallista SCA6:ssa. Lihasvoimat säilyvät hyvin.
Puheen puuroutuminen ja artikulaation ongelmat kuuluvat oirekuvaan. Puheääni muuttuu voimattomaksi. Nielemisvaikeutta on kuvattu. Silmien liikehäiriöistä tavallisin on silmänvärve (nystagmus), jota esiintyy kaikilla. Kaksoiskuvia todetaan lähes joka toisella. Pään vapinaa ja huimausta on osalla sairastuneista.
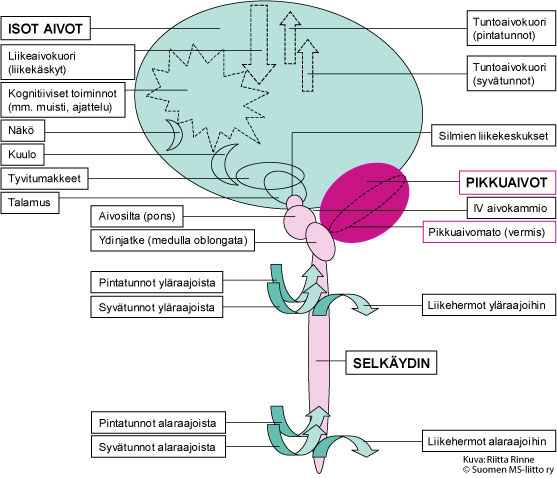
Kuva 1. SCA6:n johtuvat pikkuaivojen toimintahäiriöstä.
Harvoin todetaan jänneheijasteiden vilkastumista tai merkittävää lihasjänteyden nousua (spastisiteettia). 10%:lla on heikkenemistä nivelten asento- ja värinätunnossa. Pitkään jatkuneeseen sairauteen saattaa liittyä rakon toimintahäiriöitä.
Iäkkäillä sairastavilla henkinen suoriutuminen saattaa heiketä. Neuropsykologisia häiriöitä eli muistin, havainnoinnin, tarkkaavaisuuden tai laajemmin henkisen suorituskyvyn alenemista ei ole raportoitu. Monet potilaat ovat todenneet vaihtelua jaksamisessaan: ajoittaista väsymystä ja stressinsietokyvyn heikkenemistä esiintyy.
SCA6:n periytyminen
SCA6 periytyy autosomisesti vallitsevasti eli autosomissa dominantisti (AD). Sairautta aiheuttava geenivirhe tunnistettiin kromosomissa 19(p13) vuonna 1997. Geenitesti varmistaa SCA6:n diagnoosin. SCA6-sairauden geeni on CACNA1A ja sen tuottama valkuaisaine on kalsiumkanavan rakenneosa – P/Q-kalsiumkanavan α-1A-alayksikkö. SCA6-sairaudessa geenimutaatio on geenin luentakehyksen sisäinen ylipitkä, epävakaa CAG-jakso. Normaalissa CACNA1A-geenissä on < 19 CAG-jaksoa. SCA6-sairauden geenissä on yli > 19 CAG-jaksoa. Tavallisin SCA6-sairaudessa todettava CAG-jakson pidentymä on 22. CAG-jakso ohjaa CACNA1A-valkuaisaineen aminohappo glutamiinin syntymisen. Seurauksena on ylipitkä glutamiiniketju (PolyQ-ketju) kalsiumkanavan alayksikössä α-1A:ssa (Kuva2.).
Ylipitkä CAG-jakso on tavanomaisesti epävakaa ja jaksolla on taipumusta pidentyä seuraavassa sukupolvessa. SCA6-sairauden CAG-jakso on todettu suhteellisen stabiiliksi. Tästä johtuen sairauden varhaistumista seuraavassa sukupolvessa (antisipaatiota) ei pääsääntöisesti tapahdu. SCA6-geeni todetaan poikkeuksellisen usein henkilöllä, jonka sukulaisilla ei ataksiasairautta ole (sporadinen esiintyminen).
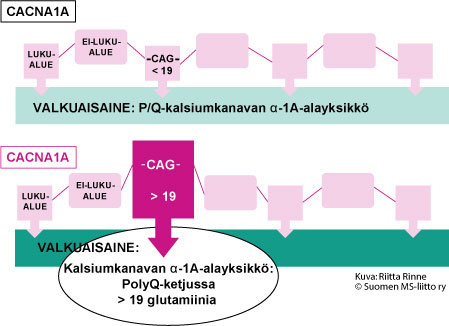
Kuva 2. SCA6-sairaudessa geenivirhe aiheuttaa kalsiumkanavan valkuaisaineeseen α-1A ylipitkän PolyQ-ketjun syntymisen.
DNA-testi varmistaa SCA6:n diagnoosin. DNA-testiä voidaan käyttää myös sairautta ennakoivana (prediktiivinen geenitesti). Määritys voidaan tehdä halutessa täysi-ikäisiltä oireettomilta, riskissä olevilta sukulaisilta tai sikiönäytteestä.
SCA6:n diagnoosi ja erotusdiagnoosi
SCA6-diagnoosiin ja erotusdiagnoosiin tarvitaan neurologinen tutkimus sekä sen perusteella arvioitavat laboratorio-, kuvantamis- ja muut hermotoimintojen tutkimukset. Muiden erikoisalojen konsultaatiot ovat usein perusteltuja. Perinnöllisyyslääkärin konsultaatio ja neuvonta sairastuneelle ja hänen sukulaisille on aina tarpeen.
Laboratoriotutkimukset verinäytteestä, lukuun ottamatta diagnostista DNA-testiä, ovat normaalit, samoin selkäydinnestenäyte (likvor-tutkimus). Magneettitutkimuksessa surkastumaa nähdään pikkuaivoissa. Muutoin aivojen rakenne on normaali. EEG-tutkimuksessa voidaan todeta purkauksellista aktiviteettia. SCA6-potilaiden aivojen aineenvaihduntaa on tutkittu positroniemissio-tutkimuksella (PET-tutkimus). Alentunutta sokeriaineenvaihduntaa on pikkuaivojen alueella, mutta aineenvaihduntamuutokset eivät rajoitu pelkästään pikkuaivoihin.
Erotusdiagnoosissa tärkein ryhmä on muut hitaasti etenevät ja ”puhtaasti” pikkuaivo-oireita aiheuttavat polyglutamiiniataksiat (ADCA III), erityisesti SCA5, SCA8, SCA11, SCA15, SCA26 ja SCA28. Suomessa tavalliseksi osoittautunut SCA8 on erityisesti muistettava erotusdiagnostiikassa. SCA6-sairaus todetaan poikkeuksellisen usein myös silloin, kun suvussa ei ataksiasairautta ole aikaisemmastaan todettu. Nämä potilaat edustavat mahdollisesti uusia SCA6-mutaatioita. Toisena vaihtoehtona on, että vanhemman lieväoireinen tai mahdollisesti vasta vanhuusiässä oirehtimaan alkanut sairaus ei ole johtanut diagnoosiin. SCA6 voi perinnöllisyydeltään muistuttaa myös autosomisesti peittyvästi periytyvää sairautta.
CACNA1A-geenissä on muitakin sairautta aiheuttavia mutaatioita eli SCA6:lle alleelisia sairauksia. Nämä sairaudet kuuluvat SCA6:n erotusdiagnoosiin. CACNA1A-geenimutaatioista johtuvia alleelisia sairauksia ovat:
- SCA6-ataksiaa muistuttava autosomisesti vallitsevasti periytyvä ataksia (CACNA1A-geenin missense mutaatio G293R), jonka oirekulku on huomattavasti SCA6:n oireita nopeampi.
- Episodinen ataksia tyyppi 2 (EA2), jonka oireille (tasapainovaikeus, huimauskohtaukset ja silmänvärve) on tyypillistä kohtauksellisuus. EA2-sairauden aiheuttaa useamman tyyppiset pistemutaatiot CACNA1A-geenissä. EA2 periytyy autosomisesti vallitsevasti. Vuosien myötä sairastuneiden oireet voivat muuttua pysyviksi ja oirekuva tällöin muistuttaa SCA6-sairautta.
- Familiaalinen hemipleeginen migreeni (FHM) aiheutuu CACNA1A-geenin missense mutaatioista ja periytyy autosomisesti vallitsevasti.
SCA6:n esiintyminen
SCA6-ataksia on Suomessa erittäin harvinainen. SCA6:lla on taipumus kasautua tietyille alueille, mikä viittaa vahvasti SCA6-geenimutaation perustajavaikutukseen. Geenin perustajavaikutuksella tarkoitetaan geenimutaation alkuperän juontamista yhteiseen kantavanhempaan. Saksassa useimmat SCA6-perheet asuvat maan luoteisosissa aivan Ranskan rajalla. Muualla Saksassa on diagnostisoitu vain yksittäisiä SCA6-perheitä. Ranskassa SCA6-geenimutaatio on äärimmäisen harvinainen kattaen vain 1–2 % maan SCA-sairauksista. Japanissa on todettu samantyyppinen SCA6-perheiden kasaantuminen tietyille alueille. SCA6 on kaikkialla maailmassa harvinainen. SCA6 edustanee noin 5–15 % tunnistetuista SCA-sairauksista. Tietyillä alueilla Japanissa peräti 31 %:lla SCA-perheistä todetaan SCA6-mutaatio.
SCA6:n esiintyminen ja oireiden alkaminen maittain
Lähteet: Baloh RW and Jen JC, 2000 ja Jiang H et al., 2005.
- Japani: SCA-perheitä 64, SCA6-perheitä 20 (31 %), SCA6-oireiden alku (vaihtelu): 52 ± 13 v (28–73 v.)
- Ranska: SCA-perheitä 74, SCA6-perheitä 1 (1 %), SCA6-oireiden alku (vaihtelu): 45 ± 14 v (24–67 v.)
- Saksa; SCA-perheitä 77, SCA6-perheitä 17 (22 %), SCA6-oireiden alku (vaihtelu): 53 ± 11 v (30–71 v.)
- USA: SCA-perheitä 75, SCA6-perheitä 9 (12 %), SCA6-oireiden alku (vaihtelu): 51 ± 6 v
- Kiina: SCA-perheitä 120, SCA6-perheitä 4 (3,3 %), SCA6-oireiden alku (vaihtelu): 45 ± 7 v (38–59 v.)
SCA6:n aiheuttamat hermovauriot
SCA6:n neuropatologiset muutokset paikallistuvat pikkuaivoihin. Vähäistä surkastumaa (atrofiaa) on myös aivorungon oliva-tumakkeissa. Mikroskooppisessa tutkimuksessa nähdään pikkuaivoissa Purkinjen solujen katoa, kohtalaista granule-solujen ja dentate-tumakkeen tuhoa sekä vähäisiä tai kohtalaisia rappeutumamuutoksia oliva-tumakkeissa.
SCA6:n hoito ja kuntoutus
SCA6:n hoitoon ei ole käytettävissä oireiden etenemistä hillitsevää tai sairautta parantavaa lääkehoitoa. SCA6:n alleelisen sairauden EA2:n hoidossa on menestyksellisesti käytetty asetazolamiidia. Vaikutusmekanismiltaan asetazolamiidi on karbonianhydraasin estäjä. Asetazolamiidia (Diamox®) on kokeiltu myös SCA6:ssa. Osalla oireissa on todettu lievittymistä hoitoannoksilla 500–750 mg vuorokaudessa. Sivuvaikutuksena on ollut raajojen tuntohäiriöitä, jotka yleensä lievittyvät ajan myötä. Pitkäaikaishoidossa asetazolamidin sivuvaikutuksena ovat munuaiskivet, joiden kehittymistä ehkäistään sitrusjuomilla. Sulfa-allergiset henkilöt voivat saada allergiaoireita. Todennäköisesti asetazoamiidi ei estä hermojen tuhoutumista. Vapinaa lievittävät lääkkeet eivät juuri auta pikkuaivoperäiseen vapinaan. Vitamiinilisiä on suositeltu silloin, kun tavanomaisesta ravinnosta niitä ei todennäköisesti riittävästi kerry.
Kuntoutustarpeen arvioiminen ja kirjaaminen kuntoutussuunnitelmaan ja seuranta ovat keskeinen osa SCA6-sairauden hyvää hoitoa. Kuntoutussuunnitelmassa on arvioitava eri terapioiden tarve, tarvittavat apuvälineet ja muut selviytymistä tukevat keinot. Tarvittavat palvelut kirjataan palvelusuunnitelmaan.
Liikuntakyvyn säilymistä tuetaan fysioterapialla. Tasapainon, lihasten hallinnan sekä koordinaation harjoittaminen omaehtoisesti kannattaa. Soveltuvista liikunnan muodoista saa ohjeita fysioterapeutilta, samoin kuin opastusta lihashuollosta ja rentoutuksesta. Puheterapeutin arviot ja ohjeet ylläpitävät kommunikaatiotaitoja. Niin kommunikaatiota, arkipäivän toimien suorittamista kuin myös liikkumista on mahdollista helpottaa apuvälinein. Sairauden aiheuttaessa lisääntyvää avun- ja hoivan tarvetta on tehtävä palvelusuunnitelma. Kuntoutussuunnitelman tavoin myös palvelusuunnitelman ajantasaisuus on sovitusti tarkistettava.
SCA6:n perustutkimuksesta
SCA6:n perustutkimuksessa selvitetään CACNA1A-geenimutaation vaikutuksia kalsiumkanavan toimintaan. CACNA1A-geeni ohjaa Ca2+-kanavan a-1A-jänniteriippuvaisen rakenneyksikön syntymistä. Kalsium-kanava on toiminnallisesti ja rakenteellisesti monimutkainen. Patologinen α-1A-rakenne voi vaikuttaa suoraan heikentävästi kalsiumkanavan läpäisyyn tai sairauden kehittymiseen voi olla monia muita tärkeitä syitä. a-1A-yksikön sisältäviä kalsium-kanavia esiintyy keskushermoston soluissa ja erityisesti pikkuaivoissa Purkinje- ja granule-soluissa.
Kirjoittaja
Riitta Rinne, LL, neurologian erikoislääkäri, Maskun neurologinen kuntoutuskeskus
Julkaistu: 1.11.2002. Päivitetty: 16.6.2006
- Kirjallisuusviitteet
- Geschwind DH et al., Spinocerebellar ataxia type 6. Frequency of the mutation and genotype‐fenotype
correlations. Neurology 49: 1247–1251, 1997 - Ophoff RA et al., Familial hemiplegic migraine and episodic ataxia type‐2 are caused by mutations in the
Ca2+ channel gene CACNL1A4. Cell 87: 543–552, 1996 - Gomez CM et al, Spinocerebellar ataxia type 6: gaze‐evoked and vertical nystagmus, Purkinje cell
degeneration, and variable age of onset. Ann Neurol 42: 933–950, 1997 - Ikeuchi T et al., Spinocerebellar ataxian type 6: CAG repeat expansion in 1A voltage‐dependent calsium
channel gene and clinical variations in Japanese population. Ann Neurol 42: 879–884, 1997 - Kaakkola S ja Rinne R: Ataksiat ja niiden erotusdiagnostiikka (katsaus). Duodecim 113: 1773–1782, 1997
- Zhuchenko O et al., Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine
expansions in the alpha 1A‐voltage‐dependent calcium channel. Nat Genet 15: 62–67, 1997 - Yue Q et al., Progressive ataxia due to a missense mutation in a calcium‐channel gene. Am J Hum Genet 61:
1078–1087, 1997 - Klockgether T et al., The natural history of degenerative ataxia: a retrospective study in 466 patients. Brain
121: 589– 600, 1998 - Moseley ML et al., Incidence of dominant spinocerebellar and Friedreich triplet repeats among 361 ataxia
families. Neurology 51: 1666–1671, 1998 - Dichgans M et al., Spinocerebellar ataxia type 6: Evidence for a strong founder effect among German
families. Neurology 52: 849–851, 1999 - Baloh RW and Jen JC: Episoidic ataxia type 2 and spinocerebellar ataxia type 6. Kirjassa: Handbook of Ataxia Disorders, 447–467. Ed. Klockgether T, Marcel Dekker Inc., New York‐Basel, 2000
- Schöls L et al., Genetic background of apparently idiopathic sporadic cerebellar ataxia. Hum Genet 107: 132–137, 2000
- Storey E et al., Frequency of spinocerebellar ataxia types 1, 2, 3, 6, and 7 in Australian patients with
spinocerebellar ataxia. Am J Med Genet 95: 351–357, 2000 - Takeichi N et al.,: Dissociation of smooth pursuit and vestibulo‐ocular reflex cancellation in SCA‐6.
Neurology 54: 860–866, 2000 - Tang B et al., Frequency of SCA1, SCA2, SCA3/MJD, SCA6, SCA7, and DRPLA CAG trinucleotide repeat
expansions in patients with hereditary spinocerebellar ataxia from Chinese kindreds. Arch Neurol 57: 540–544, 2000 - Ishikawa K et al., Cytoplasmic and nuclear polyglutamine aggregates in SCA6 Purkinje cells. Neurology 56:
1753–1756, 2001 - Koh SH et al., Spinocerebellar atxia type 6 and episodic ataxia type 2 in a Korean family. J Korean Med Sci
16: 809–813, 2001 - Mariotti C et al., Pathogenic effect of an intermediate size SCA‐6 allele (CAG)19 in a homozygous patient.
Neurology 57: 1502–1504, 2001 - Piedras‐Renteira ES et al., Increased expression of α‐1A Ca2+ channel currents arising from expanded
trinucleotide repeats in spinocerebellar ataxia type 6. J Neurosci 21: 9185–9193, 2001 - Sinke RJ et al., Clinical and molecular correlations in spinocerebellar ataxia type 6. A study of 24 Dutch
families. Arch Neurol 58: 1839–1844, 2001 - Soong B et al., Metabolic characterization of a spinocerebellar ataxia type 6. Arch Neurol 58: 300–304,
2001 - Inagaki A et al., Positron emission tomography and magnetic resonance imaging in spinocerebellar ataxia
type 2: a study of symptomatic and asymptomatic individuals. Eur J Neurol 12: 725–728, 2005 - Jiang H et al., Frequency analysis of autosomal dominant spinocerebellar ataxias in mainland Chinese
patients and clinical and molecular characterization of spinocerebellar ataxia type 6. Chin Med J 118: 837–843, 2005 - Juvonen V et al. The occurence of dominant spinocerebellar ataxias among 251 Finnish ataxia patients and
the role of predisponding large normal alleles in a genetically isolated population. Acta Neurol Scand 111:
154–1562, 2005 - Mascke M et al., Clinical feature profile of spinocerebellar ataxia type 1 – 8 predicts genetically defined
subtypes. Mov Disord 20: 1405–1412, 2005 - van de Warrenburg BPC et al., Age at onset variance analysis in spinocerebellar ataxias: a study in a Dutch‐
Frech cohort. Ann Neurol 57: 505–512, 2005 - Wüllner U et al., Dopamine transporter positron emission tomography in spinocerebellar ataxias type 1, 2,
3 and 6. Arch Neurol 62: 1280–1285, 2005
- Geschwind DH et al., Spinocerebellar ataxia type 6. Frequency of the mutation and genotype‐fenotype
Järjestötoiminta
Potilasjärjestö: Neuroliitto ry; ataksiaverkosto
Muita tietolähteitä
- The National Ataxia Foundation: www.ataxia.org
- Sveriges socialstyrelsens kunskapsdatabas om ovanliga diagnoser innehåller information om fler än 300 ovanliga sjukdomar och tillstånd.