
Spinoserebellaarinen ataksia tyyppi 2 (SCA2)
- ICD-10: G11.8 Muut määritetyt perinnölliset ataksiat, OMIM: 183090, ORPHA: 98756
- Muut sairaudesta käytetyt nimet:
- Spinoserebellaarinen atrofia II
- Olivopontoserebellaarinen atrofia, Holguinin tyyppi
- Olivopontoserebellaarinen atrofia 2
- Spinoserebellaarinen ataksia, Kuubalainen tyyppi
- Serebellaarinen degeneraatio ja hitaat silmien liikkeet
- Wadia-Swami oireyhtymä
- Spinoserebellaarinen degeneraatio ja hitaat silmien liikkeet; SDSEM
Spinoserebellaarinen ataksia tyyppi 2 (SCA2) on pikkuaivojen ja niiden hermoratayhteyksien toimintahäiriöihin johtava etenevä sairaus. SCA2 periytyy autosomisesti vallitsevasti eli dominantisti (AD).
Oireiden alkaminen ja niiden eteneminen vaihtelevat sairastuneilla merkittävästi. Pikkuaivo-oireiden (tasapainovaikeus, hienomotoriikan heikkeneminen ja puheen sammallus) lisäksi SCA2:ssa todetaan hitaat silmien seurantaliikkeet sekä tuntojen alenemaa raajoissa (polyneuropatia). Sairauteen voi liittyä kognitiivisia muutoksia ja osalle kehittyy dementia. Sairautta aiheuttava geenimutaatio tunnetaan ja sen määrittäminen verinäytteestä varmistaa SCA2-diagnoosin. SCA2 lukeutuu polyglutamiiniataksioihin (PolyQ-ataksiat).
SCA2 alkaa tavallisimmin keski-ikäisenä (30–40 vuoden iässä), mutta sairaus voi alkaa jo huomattavasti varhemmin. Nuorin SCA2-diagnoosin saanut on ollut 6 kuukauden ikäinen ja vanhin lähes 80 vuoden ikäinen. Sairauden kestossa on myös huomattavaa vaihtelua (1–30 v; km. 17 vuotta). Liikuntavaikeudet johtavat apuvälineiden tarpeeseen km. 10–20 vuoden kuluttua ensioireista. SCA2 lyhentää elinikää – km. kuolinikä on hieman yli 60 vuoden ikäisenä. Toistaiseksi selvittämättömästä syystä SCA2-sairauden on todettu etenevän naisilla nopeammin. Aikuisiällä alkavassa SCA2:ssa tasapaino- ja kävelyvaikeudet ovat lähes poikkeuksetta sairauden ensioireet (Kuva 1).
Aikuisiän SCA2:n taudinkuvaan kuuluvat:
- Tasapainon ja kävelyn epävarmuus (kävelyataksia)
- Vartaloataksia ja liikkeiden hallintavaikeus
- 2/3:lla todetaan alaraajojen alentunut lihasjänteys (hypotonia) ja jänneheijasteiden vaimeneminen tai sammuminen
- Yläraajaheijasteiden vaimeneminen ja sammuminen (hypo- ja arefleksia)
- Hidastuneet silmäliikkeet todetaan 2/3:lla potilaista; tyypillistä on katseen kohdistamisen ongelma ja seurantaliikkeiden hitaus (hypometriset sakkadit)
- Puhenopeuden hidastuminen ja artikulaation epäselvyys (dysartria)
- Hienomotoriikan epätarkkuus
- Raajojen kärkiosien tuntoheikkous
Lisäksi todetaan vaihtelevasti:
- nielemisen ongelmia (dysphagia)
- alaraajojen lihaskramppeja
- kielen ja suun alueen lihasnykäyksiä (faskikulaatioita ja myokymiaa)
- pakkoliikkeitä (dystoniaa ja koreaa)
- heikentynyt värinätunto (vibraatiotunto)
- yläraajojen vapinaa (tremor)
- rakon toimintahäiriöitä
- keskittymisongelmia ja toimintojen suunnittelun ja toteuttamisen ongelmia (kognitiivisia muutoksia) sekä dementiaa
- SCA2:een liittyy harvoin lihasjänteyden nousua (spastisiteettia) ja heijasteiden vilkastumista (hyperrefleksiaa)
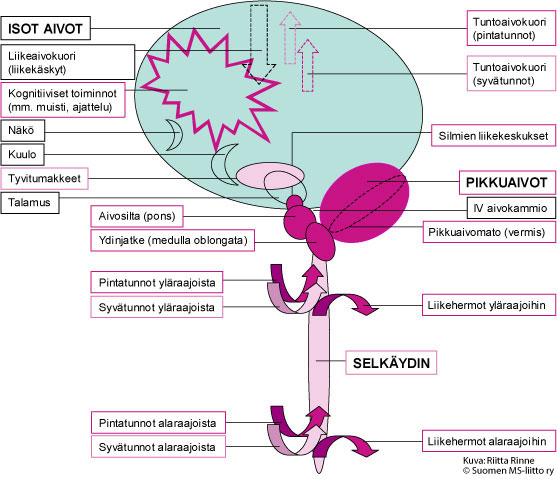
Kuva 1. Aikuisiällä alkavan SCA2:n aiheuttama neurologinen oireisto johtuu pikkuaivojen ja niiden hermoratayhteyksien toiminnan heikkenemisestä. Kuvassa on tummalla violetilla merkitty ne hermoston alueet, joissa SCA2-sairaus aiheuttaa aina muutoksia ja vaaleammalla violetilla ne hermoston alueet, joissa muutoksia voi esiintyä.
SCA2:n taudinkuva ja oireiden eteneminen
Kaikilla SCA2 potilailla todetaan tasapainon epävarmuus ja liikkumisen vaikeudet. Hienoinen tasapainohaitta voidaan havaita rappuja laskeutuessa tai horjahteluna käännöksissä. Ongelmat lisääntyvät hämärässä ja pimeässä. Sairauden edetessä seisomatasapaino heikkenee. SCA2:ssä todetaan myös vartalon hallintavaikeus: istuma-asento horjahtaa herkästi ja kurkottelut ovat hankalia.
Käsinäppäryys hidastuu. Työskentelyä haittaa käsivapina (aktion tremor). Monimutkaisten liiketaitojen – esim. pianokappaleiden soittaminen, koneella kirjoittaminen jne. – suorittaminen on työlästä. Käsien osumatarkkuus huononee eikä pienten esineiden keräily tahdo onnistua. Sairauden edetessä opitut taidot ”unohtuvat” eikä uusia liiketaitoja opita.
Puheen epäselvyys ja artikulaation vaikeudet kuuluvat oirekuvaan jo sairauden ensi vuosina. Puherytmi hidastuu ja äänenvoimakkuus vaihtelee. Taudin edetessä esiintyy ongelmia nielemisessä: ruoka ja erityisesti juomat menevät väärään kurkkuun.
Silmien liikehäiriöt kuuluvat SCA2:n taudinkuvaan. Tyypillisesti todetaan silmien seurantaliikkeiden hidastuminen (hypometriset sakkadit) jo sairauden ensivuosina. Käytännössä katseen kohdistaminen haluttuun suuntaan ei onnistu ja seurauksena on pään kääntäminen katsesuuntaan. Joillakin SCA2 potilailla silmien liikehäiriöt ovat sairauden ensioireita. Silmien liikehäiriöt voivat aiheuttaa kaksoiskuvia. Silmien liikelaajuudet voivat sairauden edetessä rajoittua (oftalmoplegia). SCA2:ssa todetaan harvoin silmänvärvettä (nystagmusta). Näkö pysyy hyvänä.
Yli puolella esiintyy ääreishermoston toimintahäiriöitä. Pintatunnot (kosketus, kipu ja lämpötilan tunnistus) heikkenevät, tunnon alenema on yleensä voimakkaampi ylä- kuin alaraajoissa. Lihasvoiman aleneminen ja lihasjänteyden heikkeneminen (hypotonia) kuuluvat oirekuvaan sairauden ensi vuosista. Värinä- ja asentotunnon heikkeneminen todetaan sairauden edetessä käytännössä kaikilla. Rakon toimintahäiriöitä on todettu jopa 70 %:lla.
SCA2:ssä todetaan vaihtelevasti oireita, jotka johtuvat toimintahäiriöistä laajemminkin keskushermoston ja ääreishermoston alueella. Osalla potilaista todetaan pakkoliikkeitä: dystoniaa ja koreaa. Dystonia on raajojen kiertävää, vääntävää pakkoliikettä tai pään tahatonta kääntymistä. Korea-pakkoliikkeet ovat tahattomia, hieman matomaisia, epärytmisiä nykäyksiä. Niin dystonia kuin korea tulevat herkimmin esille niskan, hartioiden ja pään alueella sekä yläraajoissa. Yksittäisten lihasten tai pienten lihasryhmien nykinöitä (faskikulaatioita ja myokymiaa) havaitaan erityisesti kasvoissa ja kielessä.
SCA2:een liittyy kognitiivisia ongelmia. Kognitiivisia muutoksia todetaan sekä nuorena sairastuneilla että pitkään sairastaneilla. Kognitiivisten muutosten esiintyminen ei ole yhteydessä SCA2:ta aiheuttavan geenivirheen (CAG-jakson pidentyminen) CAG-jakson monistumien määrään. Ne eivät myöskään korreloin sairauden aiheuttaman liikehaitan kanssa. SCA2:n kognitiiviset muutokset haittaavat toimintojen aloittamista, niiden suunnittelua ja loppuunsaattamista. Kognitiiviset ongelmat voivat edetä arkipäivän selviytymistä heikentäviksi eli dementiaksi (25–33 %:lla).
SCA2:n periytyminen
SCA2 periytyy autosomisesti vallitsevasti eli autosomissa dominantisti (AD). Sairautta aiheuttava geenivirhe tunnistettiin kromosomissa 12(q23.1) vuonna 1996. Geenitesti varmistaa SCA2:n diagnoosin. SCA2-sairauden geeni on ATXN2 ja sen tuottama valkuaisaine on Ataksiini – 2. SCA2-sairaudessa geenimutaatio on geenin luentakehyksen sisäinen ylipitkä, epävakaa CAG-jakso (dynaaminen mutaatio). Normaalissa ATXN2-geenissä on alle 32 CAG-jaksoa. SCA2-sairauden geenissä on yli 32 CAG-jaksoa. CAG-nukleotidikomikko ohjaa aminohappo glutamiinin syntymisen. Seurauksena on ylipitkä glutamiiniketju (PolyQ-ketju) Ataksiini -2:ssä (Kuva2.)
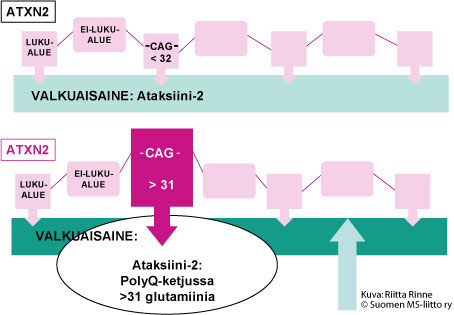
Kuva 2. SCA2-sairaudessa geenivirhe aiheuttaa Ataksiini-2 valkuaisaineeseen ylipitkän PolyQ-ketjun syntymisen.
Ylipitkän CAG-jakson epävakaus (dynaaminen mutaatio) merkitsee CAG-jakson taipumusta pidentyä seuraavassa sukupolvessa. CAG-jakson pituuden ja oireiden puhkeamisen välillä SCA2:ssa on todettu käänteistä korrelaatiota: pitkä CAG-jakso on yhteydessä oireiden varhaiseen puhkeamiseen ja nopeaan taudinkulkuun (Lista genotyyppi–fenotyyppi-korrelaatiosta alla). Sairauden varhaistumista seuraavassa sukupolvessa kutsutaan antisipaatioksi. CAG-jakson pidentymistaipumus on havaittu etenkin sairauden periytyessä isältä. Yli 45 CAG-jaksoa sisältävä ATXN2-geeni aiheuttaa sairastumisen yleensä alle 20 vuoden ikäisenä. Sen sijaan 32–40 CAG-jaksoa sisältävä ATXN2-geeni aiheuttaa sairauden ilmenemisen yleensä vasta yli 30 vuoden ikäisenä. On todettu, että sisarussarjassa samanpituisen CAG-jakson omaavat voivat sairastua eri-ikäisinä, eivätkä sisarusten oireet etene samaan tahtiin. CAG-jakson pituus ei siis yksinomaisesti määrää SCA2-sairauden oirekulkua.
Genotyyppi–fenotyyppi-korrelaatiota SCA2:ssa
- Genotyyppi – CAG-jaksoja > 200, oireiden alkaminen: imeväisikäisenä, tyypillistä oirekuvassa (fenotyyppi): Hypotonia, hengityksen heikkous, imemisvaikeus, viivästynyt motorinen ja henkinen kehitys
- Genotyyppi – CAG-jaksoja 45 tai > 45, oireiden alkaminen: < 20 v., tyypillistä oirekuvassa (fenotyyppi): Dystonia, myoklonia, myokymia ja ataksia
- Genotyyppi – CAG-jaksoja 35–44, oireiden alkaminen: 20–60 v., tyypillistä oirekuvassa (fenotyyppi): Ataksia (alku km. 27 ± 11 v), sensomotorinen polyneuropatia ja Parkinsonin taudin kaltaiset oireet (alku km. 46 ± 14 v)
- Genotyyppi – CAG-jaksoja 33–34, oireiden alkaminen: > 60 v., tyypillistä oirekuvassa (fenotyyppi): Sensomotorinen polyneuropatia ja ataksia
SCA2:n diagnoosi varmistuu DNA-testillä
DNA-testi varmistaa SCA2:n diagnoosin. DNA-testiä voidaan käyttää myös sairautta ennakoivana (prediktiivinen geenitesti). Määritys voidaan tehdä halutessa täysi-ikäisiltä oireettomilta, riskissä olevilta sukulaisilta tai sikiönäytteestä.
SCA2:n diagnoosi ja erotusdiagnoosi
SCA2-sairauden diagnoosiin ja erotusdiagnoosiin tarvitaan neurologinen tutkimus sekä sen perusteella arvioitavat laboratorio-, kuvantamis- ja muut hermotoimintojen tutkimukset. Muiden erikoisalojen konsultaatiot ovat usein perusteltuja. Perinnöllisyyslääkärin konsultaatio ja neuvonta sairastuneelle ja sukulaisille on aina tarpeen.
Laboratoriotutkimukset verinäytteestä, lukuun ottamatta diagnostista DNA-testiä, ovat normaalit, samoin selkäydinnestenäyte (likvor-tutkimus). Magneettitutkimuksessa todetaan hermokudoskatoa etenkin pikkuaivoissa ja aivorungossa (olivopontoserebellaarinen atrofia). Neurofysiologisissa tutkimuksissa tavanomainen löydös on aksonaalinen tuntohermoihin painottuva polyneuropatia, joka voi olla merkittävämpää yläraajoissa. Aivojen aineenvaihduntaa kuvaavassa positroni emissio tomografiassa (PET-tutkimus) on todettu alentunutta sokeriaineenvaihduntaa pikkuaivoissa, aivorungossa ja isoissa aivoissa parietaalisella korteksilla. Dopamiiniaineenvaihdunnan puutetta oli todettavissa tyvitumakkeiden striatumissa. Tyvitumakkeiden dopamiinin sitominen poikkesi Parkinsonin taudin muutoksista.
Erotusdiagnoosissa tärkein ryhmä on muut PolyQ-ataksiat, erityisesti SCA1 ja SCA3. Periaatteessa myös muut SCA-sairaudet, joissa oireet eivät rajoitu puhtaasti pikkuaivo-oireisiin (ADCA I), tulevat kyseeseen. Perinnöllinen, nuorella iällä alkava Parkinsonin tauti ja perinnölliset polyneuropatiat (HSMN-sairaudet) ovat erotusdiagnoosissa muistettavia vaihtoehtoja. SCA2 todetaan äärimmäisen harvoin sporadisena eli henkilöllä, jonka suvussa ei aikaisemmastaan ole esiintynyt eteneviä neurologisia sairauksia.
SCA2:n esiintyminen
SCA2-diagnooseja Suomessa on todettu vähän. Tavallisin SCA-sairauksista SCA2 on Meksikossa, Intiassa, Italiassa ja Englannissa. Näissä maissa vaihtelevasti 25–57 % SCA-diagnooseista on ATXN2-geenimutaatiosta johtuvia. Suomen ohella Portugalissa, Koreassa, Brasiliassa ja Australiassa SCA2 on erittäin harvinainen.
SCA2:n aiheuttamat hermovauriot
Neuropatologisessa tutkimuksessa aivojen paino on vähentynyt. Surkastumista (atrofiaa) on pikkuaivoissa, aivorungossa, selkäytimessä ja isoaivojen otsalohkoissa. Mikroskooppisessa tutkimuksessa nähdään huomattava pikkuaivojen Purkinjen solujen määrän väheneminen sekä solukatoa alempien oliva-tumakkeiden että ponsin alueella. Pontoserebellaarisissa ja spinoserebellaarisissa hermoratayhteyksissä on atrofiaa. Lähes kaikilla on selkäytimen takajuosteissa rappeutumaa. Aivojen tyvitumakkeissa solukatoa todetaan erityisesti substantia nigrassa.
SCA2:n hoito ja kuntoutus
Ataksian lievittäminen käytössä olevin lääkkein ei onnistu. Vapinaoireeseen voi yrittää beeta-salpaajaa. Parkinsonin taudin lääkehoidossa käytettävät L-dopamiini tai dopamiinin vaikutusta tehostavat valmisteet (dopamiini-agonistit) voivat lievittää SCA2-sairauteen liittyviä Parkinsonin sairauden kaltaisia oireita ja jalkojen levottomuutta. Pirasetaami (12–18 g/vrk) on raportoitu tehokkaaksi myoklonuksen lievittämisessä. Rakon toimintahäiriöihin on niin ikään saatavissa apua. Aloitettavan oirelääkkeen tehon arvioinnissa on oltava kriittinen. Etenevässä sairaudessa lääkkeen tarpeellisuutta ja sen annosta on syytä tarkistaa. Hoitava lääkäri antaa ohjeet annosmäärän säätelyyn. Pitkään käytössä olleiden keskushermostoon vaikuttavien lääkkeiden lopettamista ei saa tehdä äkillisesti.
Kuntoutustarpeen arvioiminen ja kirjaaminen kuntoutussuunnitelman ja seuranta ovat keskeinen osa SCA2-sairauden hyvää hoitoa. Kuntoutussuunnitelmaan on kirjattava eri terapioiden tarve, tarvittavat apuvälineet ja muut selviytymistä tukevat keinot. Tarvittavat palvelut kirjataan palvelusuunnitelmaan.
Liikuntakyvyn säilymistä tuetaan fysioterapialla. Tasapainon, lihasten hallinnan sekä koordinaation harjoittaminen omaehtoisesti kannattaa. Soveltuvista liikunnan muodoista saa ohjeita fysioterapeutilta, samoin kuin opastusta lihashuollosta ja rentoutuksesta. Puheterapeutin arviot ja ohjeet ylläpitävät kommunikaatiotaitoja ja keinoja on löydettävissä myös turvalliseen nielemiseen. Nielemisvaikeuksien takia ruoka voi mennä ”väärään kurkkuun” eli henkitorveen (aspiraatio). Aspiraatio altistaa toistuville keuhkotulehduksille ja pahimmillaan tukehtumiselle. Aspiraation riski tutkitaan videofluorometrialla. Nielemisen turvallisuuteen voidaan vaikuttaa. Nielemisen koordinointiongelmissa on vältettävä karkeusasteeltaan vaihtelevia ruokia. Kiinteä ruoka ja nesteet on nautittava erikseen. Nielemisen turvallisuutta lisäävät ruuan ja nesteiden tasalaatuisuus ja ruokailun rauhoittaminen. Vatsanpeitteiden läpi asetettavaa PEG-letkua suositellaan silloin, jos nieleminen aspiraatiovaaran takia ei ole turvallista.
Niin kommunikaatiota, arkipäivän toimien suorittamista kuin myös liikkumista on mahdollista helpottaa apuvälinein. Sairauden aiheuttaessa lisääntyvää avun- ja hoivan tarvetta on tehtävä palvelusuunnitelma. Kuntoutussuunnitelman tavoin myös palvelusuunnitelman ajantasaisuus on sovitusti tarkistettava.
SCA2:n perustutkimuksesta
SCA2-sairauden tutkimukseen on kehitetty hiirimalleja. ATXN2-geeniä esiintyy laajalti hermostossa kuin myös muissa kudoksissa lukuun ottamatta keuhkoja ja munuaisia. Keskushermossa Ataksiini-2 paikantuu pikkuaivojen Purkinjen soluihin, hippokampuksen pyramidineuroneihin, tyvitumakkeiden substantia nigraan sekä trochlearis-hermon tumakkeisiin. Niin poikkeavan kuin normaalinkin Ataksiini-2 valkuaisaineen on todettu sijaitsevan aina solulimassa. Poikkeava Ataksiini-2 kertyy solulimaan muodostaen saostumia (aggregaatteja). Solulimassa Ataksiini-2 paikallistuu Golgin kalvostoihin. In vitro-tutkimuksissa on patologisen Ataksiini-2:n osallistuvan solun itseohjattuun, ennenaikaiseen kuolemaan (apoptoosiin). Genotyyppi-fenotyyppikorrelaatio on ilmeinen SCA2-sairaudessa. CAG-jakson pituus ei kuitenkaan riitä yksinomaisesti selvittämään SCA2:n genotyyppi-fenotyyppikorrelaatiota. Myös muut geenit vaikuttavat oireiden puhkeamiseen ja niiden etenemiseen. Tutkimushavaintojen perusteella SCA2:n solutason toimintahäiriöt poikkeavat selvästi SCA1:ssä ja SCA3:ssa todetuista.
Kirjoittaja
Riitta Rinne, LL, neurologian erikoislääkäri, Maskun neurologinen kuntoutuskeskus
Julkaistu 14.10.2002. Päivitetty 16.6.2006.
- Kirjallisuusviitteet
- Burk K et al., Autosomal dominant cerebellar ataxia type I. Clinical features and MRI in families
with SCA1, SCA2 and SCA3. Brain 119: 1497 – 1505, 1996 - Imbert G et al., Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with high sensitivity
to expanded CAG/glutamine repeats. Nat Genet 14: 285 – 291, 1996 - Pulst SM et al., Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar
ataxia type 2. Nat Genet 14: 269 – 276, 1996 - Sanpei K et al: Identification of the spinocerebellar ataxia type 2 gene using a direct identification
of repeat expansion and cloning technique, DIRECT. Nat Genet 14: 277 – 284, 1996 - Adams C et al., Clinical and molecular analysis of a pedigree of southern Italian ancestry with
spinocerebellar ataxia type 2. Neurology 49: 1163 – 1166, 1997 - Cancel G et al., Molecular and clinical correlations in spinocerebellar ataxia 2: a study of 32
families. Hum Mol Genet 6: 709 – 715, 1997 - Geschwind DH et al., The prevalence and wide clinical spectrum of the spinocerebellar ataxia type
2 trinucleotide repeat in patients with autosomal dominant cerebellar ataxia. Am J Hum Genet 60:
842 – 850, 1997 - Kaakkola S ja Rinne R: Ataksiat ja niiden erotusdiagnostiikka (katsaus). Duodecim 113: 1773 –
1782, 1997 - Lorenzetti D et al., The expansion of the CAG repeat in ataxin‐2 is a freguent cause of autosomal
dominant spinocerebellar ataxia. Neurology 49: 1009 – 1013, 1997 - Buttner N et al., Oculomotor phenotypes in autosomal dominant ataxias. Arch Neurol 55: 1353 –
1357, 1998 - Gambardella A et al., CAG repeat length and clinical features in three Italian families with
spinocerebellar ataxia type 2 (SCA2): early impairment of Wisconsin Card Sorting Test and saccade
velocity. J Neurol 245: 647 – 652, 1998 - Grewal RP et al.: Clinical and genetic analysis of a distinct autosomal dominant spinocerebellar
ataxia. Neurology 51: 1423 – 1426, 1998 - Klockgether T and Evert B: Genes involved in hereditary ataxias. Trends Neurosci 21: 413 – 418,
1998 - Klockgether T et al., The natural history of degenerative ataxia: a retrospective study in 466
patients. Brain 121: 589 – 600, 1998 - Moseley ML et al., Incidence of dominant spinocerebellar and Friedreich triplet repeats among
361 ataxia families. Neurology 51: 1666 – 1671, 1998 - Bürk K et al., Cognitive deficits in spinocerebellar ataxia 2. Brain 122: 769 – 777, 1999
- Huynh DP et al., Expression of Ataxin‐2 in brain from normal individuals and patients with
Alzheimer`s Disease and Spinocerebellar Ataxia 2. Ann Neurol 45: 232 –241, 1999 - Storey E et al., Spinocerebellar Ataxia Type 2. Clinical features of a pedigree displaying prominent
frontal‐executive dysfunction. Arch Neurol 56: 43 – 50, 1999 - Bürk K and Dichgans J: Spinocerebellar Ataxia Type 2. Kirjassa: Handbook of Ataxia Disorders, 363 –
384. Ed. Klockgether T. Marcel Dekker Inc., New York‐Basel, 2000 - Costanzi‐Porrini S et al., An interrupted 34‐CAG repeat SCA‐2 allele in patients with sporadic
spinocerebellar ataxia. Neurology 54: 491 – 493, 2000 - Fernandez M et al., Late‐onset SCA2: 33 CAG repeats are sufficient to cause disease. Neurology 55:
569 – 572, 2000 - Gwinn‐Hardy K et al., Spinocerebellar ataxia type 2 with parkinsonism in ethnic Chinese. Neurology
55: 800 – 805, 2000 - Hayes S et al., CAG repeat length in RAI1 is associated with age at onset variability in
spinocerbellar ataxia type 2 (SCA2). Hum Mol Genet 9: 1753 – 1758, 2000 - Huynh DP et al., Nuclear localization or inclusion body formation of ataxin‐2 are not necessary for
SCA2 pathogenesis in mouse or human. Nature Genetics 26: 44 – 50, 2000 - Saleem O et al., Molecular analysis of autosomal dominant hereditary ataxias in the Indian
population: high frequency of SCA2 and evindence for common founder mutation. Hum Genet
106: 179 – 187, 2000 - Storey E et al., Frequency of spinocerebellar ataxia types 1, 2, 3, 6, and 7 in Australian patients
with spinocerebellar ataxia. Am J Med Genet 95: 351 – 357, 2000 - Tang B et al., Frequency of SCA1, SCA2, SCA3/MJD, SCA6, SCA7, and DRPLA CAG trinucleotide
repeat expansions in patients with hereditary spinocerebellar ataxia from Chinese kindreds. Arch
Neurol 57: 540 – 544, 2000 - Abele M et al., Restless legs syndrome in spinocerebellar ataxia types 1, 2, and 3. J Neurol 248: 311
– 4, 2001 - Boesch SM et al., Proton magnetic resonance spectroscopic imaging reveals differences in
spinocerebellar ataxia types 2 and 6. J Magn Reson Imaging 13: 553 – 559, 2001 - Furtado S et al., SCA‐2 presenting as parkinsonism in an Alberta family. Clinical, genetic, and PET
findings. Neurology 59: 1625 – 1627, 2002 - Huynh DP et al., Expansion of the polyQ repeat in ataxin‐2 alters its Golgi localization, disrupts the
Golgi complex and causes cell death. Hum Mol Genet 12: 1485 – 1496, 2003 - Boesch SM et al., Abnormalities of dopaminergic neurotransmission in SCA2: a combined 1231‐
betaCIT and 1231‐IBZM SPECT study. - Guerrini L et al., Brainstem neurodegeneration correlates with clinical dysfunction in SCA1 byt not
in SCA2. A quantitative volumetric, diffusion and proton spectroscopy MR study. Brain 127: 1785 –
1795, 2004 - Infante J et al., Spinocerebellar ataxia type 2 with Levodopa‐responsive parkinsonism culminating
in motor neuron disease. Mov Disord 19: 848 – 952, 2004 - Lu C‐C et al., The parkinsonian phenotype of spinocerebellar ataxia type 2. Arch Neurol 61: 35 –
38, 2004 - Morretti P et al., Spinocerebellar ataxia type 2 (SCA2) presenting with ophtalmoplegia and
developmental delay in infancy. Am J Med Genet 124: 392 – 396, 2004 - Rüb U et al., Damage to the reticulotegmental nucleus of pons in spinocerebellar ataxia type 1, 2,
and 3. Neurology 63: 1258 – 1263, 2004 - Sinha KK et al., Autosomal dominant cerebellar ataxia: SCA2 is the most frequent mutation in
Eastern India. J Neurol Neurosurg Psychiatry 75: 448 – 452, 2004 - van de Warrenburg BPC et al., Peripheral nerve involvement in spinocerebellar ataxias. Arch
Neurol 61: 257 – 261, 2004 - Gierga K et al., Involvement of the cranial nerves and their nuclei in spinocerebellar ataxia type 2
(SCA2). Acta Neuropathol 109: 617 – 131, 2005 - Inagaki A et al., Positron emission tomography and magnetic resonance imaging in spinocerebellar
ataxia type 2: a study of symptomatic and asymptomatic individuals. Eur J Neurol 12: 725 – 728,
2005 - Koeppen AH: The pathogenesis of spinoserebellar ataxia. Cerebellum 4: 62 – 73, 2005
- Mascke M et al., Clinical feature profile of spinocerebellar ataxia type 1–8 predicts genetically
defined subtypes. Mov Disord 20: 1405 – 1412, 2005 - Pulst SM et al., Sipnocerebellar ataxia type 2: polyQ repeat variation in the CACBA1A calcium
channel modifies age at onset. Brain 128: 2297 – 2303, 2005 - Rüb U et al., Spinocerebellar ataxias type 2 and 3: degeneration of the precerebellar nuclei isolates
the three phylogenetically defined regions of the cerebellum. J Neural Trasnm 112: 1523 – 1545,
2005 - Scifried C et al., Saccade velocity as a surrogate disease marker in spinocerebellar ataxia type 2.
Ann N Y Acad Sci 1039: 524 – 527, 2005 - van de Warrenburg BPC et al., Age at onset variance analysis in spinoserebellar ataxias: a study in
a Dutch‐French cohort. Ann Neurol 57: 505 – 512, 2005 - Wüllner U et al., Dopamine transporter positron emission tomography in spinocerebellar ataxias
type 1, 2, 3 and 6. Arch Neurol 62: 1280 – 1285, 2005 - Crum BA and Josephs KA: Varied electrophysiologic patterns in spinocerebellar ataxia type 2. Eur J
Neurol 13: 104 – 197, 2006 - Gonatas NK et al., Fragmentation of the Golgi apparatus in neurodegenerative diseases and cell
death. J Neurol Sci - de Rosa A et al., Suppression myoklonus in SCA2 by piracetam. Mov Disord 21: 116 – 118, 2006
- Burk K et al., Autosomal dominant cerebellar ataxia type I. Clinical features and MRI in families
Järjestötoiminta
Potilasjärjestö: Neuroliitto ry; ataksiaverkosto
Muita tietolähteitä
- The National Ataxia Foundation: www.ataxia.org
- Sveriges socialstyrelsens kunskapsdatabas om ovanliga diagnoser innehåller information om fler än 300 ovanliga sjukdomar och tillstånd.