
Polyglutamiiniataksiat eli PolyQ-ataksiat – katsaus
ICD–10: G11.8 Muut määritetyt perinnölliset ataksiat, ORPHA: 99
- Spinoserebellaarinen ataksia tyyppi 1 (SCA1)
- Spinoserebellaarinen ataksia tyyppi 2 (SCA2)
- Spinoserebellaarinen ataksia tyyppi 3 (SCA3) eli Machado-Josephin tauti (MJD)
- Spinoserebellaarinen ataksia tyyppi 6 (SCA6)
- Spinoserebellaarinen ataksia tyyppi 7 (SCA7)
- Spinoserebellaarinen ataksia tyyppi 17 (SCA17)
- Dentatorubropallidolysiaalinen atrofia (DRPLA)
Kaikissa PolyQ-ataksioissa todetaan etenevä ataksia (tasapaino-kävelyvaikeus, silmänliikehäiriöt ja puheartikulaation ongelmat). Muita neurologisia oireita todetaan vaihtelevasti. SCA6:ssa oireet tavanomaisesti rajoittuvat puhtaaseen pikkuaivoataksiaan ja voivat sairauden ensivuosina olla ajoittaisia. SCA7:ään liittyy aina näön heikkeneminen (makuladegeneraatio ja verkkokalvon rappeutuma). Kognitiivinen heikkeneminen ja dementian kehittyminen todetaan erityisesti SCA17:ssä ja DRPLA:ssa. PolyQ-ataksioissa perheenjäsenten oireisto vaihtelee huomattavasti riippuen sairausoireiden alkamisiästä.
PolyQ-ataksioiden geenimutaatio on ylipitkä CAG-jakso
Sairautta aiheuttava geenimutaatio on oleva ylipitkä kolmen nukleoditiemäksen (CAG) muodostama CAG-jakso. CAG-jakso ohjaa glutamiini-nimisen aminohapon syntymisen. Seurauksena CAG-jakson poikkeavasta pituudesta on ylipitkä glutamiiniketju (GLU-ketju) valkuaisaineessa. Polyglutamiiniataksiat periytyvät aina autosomisesti vallitsevasti (AD-periytyminen). Sairastavan perimässä on toiselta vanhemmalta saatu normaalin pituisen CAG-jakson sisältävä geeni ja toiselta vanhemmalta periytynyt ylipitkän CAG-jakson sisältävä geeni (Kuva 1).
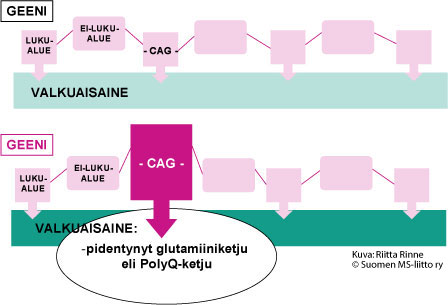
Kuva 1. Polyglutamiiniataksioissa on valkuaisaineessa poikkeava, pitkä glutamiini-aminohappojen muodostama ketju (PolyQ-ketju).
Lähes kaikki PolyQ-ataksioita aiheuttavat valkuaisaineet ovat olleet aikaisemmastaan tuntemattomia. Tästä syystä valkuaisaineet on nimetty ataksiineiksi 1, 2, 3 ja 7 SCA-sairauden numeroinnin mukaisesti. Ataksiini-3:n vaihtoehtoisena nimenä on MDJ1-proteiini Euroopassa SCA3-sukujen ja Kauko-Idässä Machado-Josephin taudin nimellä tunnetun sairauden osoittauduttua saman geenimutaation aiheuttamiksi. SCA6:n geeni ohjaa kalsium-kanavan rakenneproteiinia α-1A, josta syystä SCA6 voidaan ryhmitellä myös ionikanavasairauksiin. SCA17:ssä geeni ohjaa TATA-boxiin sitoutuvaa proteiinin (TATA-box binding protein) ja DRPLA:n geenin puolestaan tiedetään ohjaavan atropiini-1-valkuaisaineen tuotantoa.
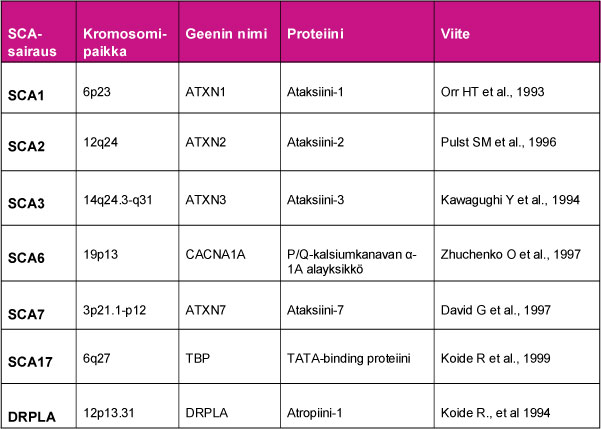
Taulu 1. PolyQ-ataksioiden geenimutaatiot, niiden kromosomisijainti sekä sairauteen liittyvät valkuaisaineet (proteiinit).
PolyQ-ataksioissa todetaan sairauden varhaistumista sukupolvittain
Luonteenomaista CAG-jakson pidentymälle on sen taipumus pidentyä sukupolvittain (dynaaminen mutaatio). PolyQ-ataksioissa ainoastaan SCA6:ssa tätä dynaamisuutta ei juuri todeta. CAG-jakson pituudella on merkitystä niin sairauden oireiden alkamiseen kuin oireiden etenemiseen. Käytännössä mitä pidempi sairautta aiheuttava CAG-jakso on, sitä varhemmin sairaus alkaa ja myös sairauden oireet etenevät suhteellisesti nopeammin. CAG-jakson monistumisherkkyys johtaa oireiden varhaisempaan alkamiseen seuraavassa sukupolvessa eli antisipaatioon. Varhain eli lapsuudessa alkavan PolyQ-sairauden oirekuva poikkeaa usein aikuisiällä alkavan ataksiasairauden oirekuvasta. CAG-jakson pituus ei yksinomaan selvitä PolyQ-sairauksissa todettavaa antisipaatiota.
PolyQ-sairauksien diagnoosi tehdään geenitestillä (DNA-testi)
SCA-sairauksien kliinisissä piirteissä on huomattavaa päällekkäisyyttä. Tästä syystä diagnoosin varmistaminen tehdään DNA-testillä. PolyQ-ataksiat alkavat keskimäärin 30–40 ikävuoden välillä, mutta lapsuusvuosina alkaneita sairauksia on todettu kaikissa muissa PolyQ-ataksioissa paitsi SCA6:ssa. SCA6:n oireet alkavat tavallisesti merkittävästi muita PolyQ-ataksioita myöhemmin noin 50 – 60 ikävuoden välillä ja oireiden eteneminen on hidasta. Tauluun 2 on koottu PolyQ-ataksioille tyypillisiä oireita sekä myös niiden erotusdiagnostiikassa huomioitavia sairauksia.
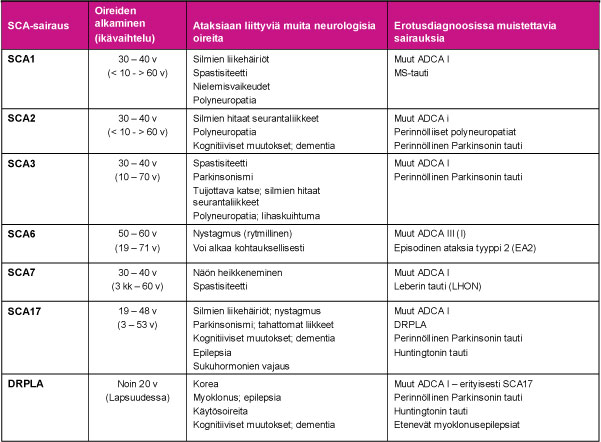
Taulu 2. Aikuisiällä alkavien PolyQ-ataksioiden kliinisiä piirteitä sekä tärkeimmät erotusdiagnoosissa muistettavat sairaudet. Kaikissa PolyQ-ataksioissa todetaan pikkuaivoataksia, kuten kaikissa SCA-sairauksissa.
PolyQ-ataksioihin ei ole parantavaa lääkehoitoa
Toistaiseksi PolyQ-ataksioiden hoito on sairauden oireiden hoitoa. SCA6 etenee hitaasti eikä lyhennä elinikää. Muissa PolyQ-ataksioissa elinennuste lyhenee. Liikunnan apuvälineitä tarvitaan noin 5–10 vuoden kuluttua oireiden alkamisesta, varhain alkaneessa sairaudessa jo aikaisemmin. Niin sairastunut kuin hänen perheensä tarvitsevat perinnöllisyysneuvonnan. Sairauden diagnoosiin käytettävällä DNA-tutkimuksella voidaan todeta oireeton sairausgeenin kantaja (ennustava eli prediktiivinen geenitesti). PolyQ-ataksian geeni voidaan DNA-testillä todeta myös sikiöstä. Oikein ajoitetulla ja suunnitelmallisella kuntoutuksella voidaan merkittävästi tukea niin sairastuneen kuin läheisten arkea.
PolyQ-ataksioiden perustutkimus
Geenimuunneltuja koe-eläimiä on kehitetty käytännössä kaikkien PolyQ-ataksioiden sairausmekanismien tutkimiseen. Koe-elämiä käytetään myös uusien hoitomahdollisuuksien tutkimisessa. Yksittäisen valkuaisaineen spesifin RNA:n ilmeneminen solussa on edellytys vastaavan DNA-molekyylin ja sen ohjaaman valkuaisaineen syntymiselle. Yhtenä hoidollisena mahdollisuutena onkin estää patologisen, ylipitkää CAG-jaksoa tuottavan RNA:n syntyä. Hoitostrategia on osoittautunut lupaavaksi ainakin SCA1-sairauden koe-eläinmallissa. Soluviljelyssä on pienimolekyylisillä aineilla yritetty estää polyglutamiinikertymien syntymistä. Alustavissa tutkimuksissa on todettu disakkaridien estävän polyglutamiini-kertymiä.
PolyQ-ataksia on suurin perinnöllisten aikuisiän ataksiasairauksien ryhmä
SCA-ataksioiden esiintyvyys maailmanlaajuisesti on 1–5 /100 000. Ne ovat harvinaisia neurologisia sairauksia. PolyQ-ataksioita esiintyy kaikkialla maailmassa ja ne edustavat ”tavallisia” SCA-sairauksia (Kuva 2). Suomessa tavallisin PolyQ-ataksia on SCA7 ja SCA1 on toiseksi yleisin PolyQ-ataksioista. Muita PolyQ-ataksioita (SCA2, SCA6 ja SCA17) on diagnosoitu harvemmin. SCA3 on maailmanlaajuisesti on tavallisin PolyQ-ataksia, mutta Suomessa sitä ei ole todettu, ei myöskään DRPLA:ta. SCA6 diagnoosi todetaan joskus henkilöillä, joiden suvussa ei muita ataksiaa sairastavia ole (sporadisena). Sporadisia SCA1, SCA2 ja SCA3 diagnooseja on ilmennyt harvoin.
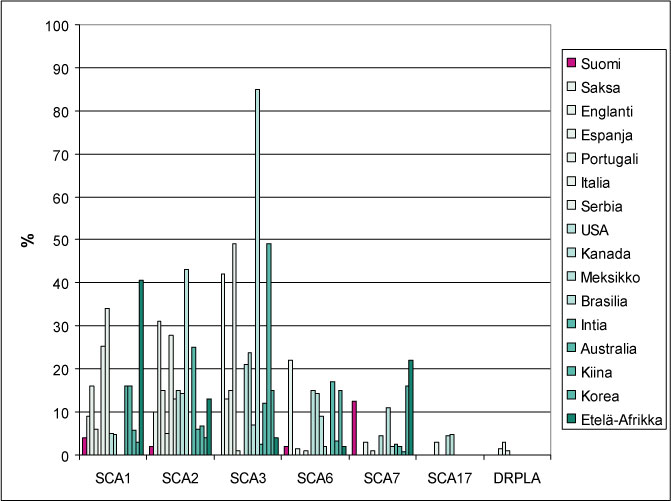
Kuva 2. PolyQ-ataksioiden esiintyminen Suomessa ja muualla.
Kirjoittaja
Riitta Rinne, LL, neurologian erikoislääkäri, Maskun neurologinen kuntoutuskeskus
Julkaistu 21.6.2006
- Kirjallisuusviitteet
- Rosenberg RN (Ed): Autosomal dominant cerebellar phenotypes: The genotype will settle the issue.
Neurology 40: 1329–1331, 1990 - Orr HT et al: Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet
4: 221–226, 1993 - Kawagughi Y et al., CAG expansions in a novel gene for Machado‐Joseph disease at chromosome 14 q 32.1.
Nat Genet 8: 213–215, 1994 - Koide R et al., Unstable expansion of CAG repeat in hereditary dentatorubralpallidoluysian atrophy
(DRPLA). Nat Genet 6: 9–13, 1994 - Rosenberg RN (Ed): Autosomal dominant cerebellar phenotypes: The genotype has settled issue. Neurology
45: 1–5, 1995 - Rosenberg RN (Ed): Spinocerebellar ataxias and ataxins. New England J Med 333 (20): 1351–1352, 1995
- Twist EC et al., Machado‐Joseph disease maps to the same region of chromosome 14 as the spinocerebellar ataxia type 3 locus. J Med Genet 32: 25–31, 1995
- Burk K et al.: Autosomal dominant cerebellar ataxia type I. Clinical features and MRI in families with SCA1,
SCA2 and SCA3. Brain 119: 1497–1505, 1996 - Pulst SM et al., Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia
type 2. Nat Genet 14: 269–276,1996 - David G et al., Cloning of the SCA7 gene reveals a highly unstable CAG‐repeat expansion Nat Genet 17: 65–70, 1997
- Kaakkola S ja Rinne R: Ataksiat ja niiden erotusdiagnostiikka (katsaus). Duodecim 113: 1773–1782, 1997
Schös L et al., Autosomal dominant cerebellar ataxia: phenotypic differences in genetically defined
subtypes? Ann Neurol 42: 924–932, 1997 - Zhuchenko O et al., Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine
expansions in the alpha 1A‐voltage‐dependent calcium channel. Nat Genet 15: 62–67, 1997
Grewal RP et al.: Clinical and genetic analysis of a distinct autosomal dominant spinocerebellar ataxia.
Neurology 51: 1423–1426, 1998 - Guinti P et al., The role of the SCA2 trinucleotide repeat expansion in 89 autosomal dominant cerebellar
ataxia families. Frequency, clinical and genetic correlates. Brain 121: 459–467, 1998 - Klockgether T and Evert B: Genes involved in hereditary ataxias. Trends Neurosci 21: 413–418, 1998
Klockgether T et al.: The natural history of degenerative ataxia: a retrospective study in 466 patients. Brain
121: 589–600, 1998 - Moseley ML et al.: Incidence of dominant spinocerebellar and Friedreich triplet repeats among 361 ataxia
families. Neurology 51: 1666–1671, 1998 - Subramony SH and Nance M: Diagnosis and management of the inherited ataxias. The Neurologist 4: 327–338, 1998
- Koide R et al., A neurological disease caused by an expanded CAG trinucleotide repeat in the TATA‐binding
protein gene: a new polyglutamine disease? Hum Mol Genet 8: 2047–2053, 1999 - Pujana MA et al., Spinocerebellar ataxias in Spanish patients: genetic analysis of familial and sporadic cases.
The Ataxia Study Group. Hum Genet 104: 516–522, 1992 - Rasmussen A et al., Molecular diagnosis of spinocerebellar ataxias in Mexican population. Am J Hum Genet
67 (Suppl 2): 342, 2000 - Schöls L et al., Genetic background of apparently idiopathic sporadic cerebellar ataxia. Hum Genet 107: 132–137, 2000
- Storey E et al., Frequency of spinocerebellar ataxia types 1, 2, 3, 6, and 7 in Australian patients with
spinocerebellar ataxia. Am J Med Genet 95: 351–357, 2000 - Tang B et al.: Frequency of SCA1, SCA2, SCA3/MJD, SCA6, SCA7, and DRPLA CAG trinucleotide repeat
expansions in patients with hereditary spinocerebellar ataxia from Chinese kindreds. Arch Neurol 57: 540–544, 2000 - Srivastava AK et al., Molecular and clinical correlation in five Indian families with spinocerebellar ataxia 12.
Ann Neurol 50: 796–800, 2001 - Subramony SH and Filla A (Ed): Autosomal dominant spinocerebellar ataxias ad infinitum? Neurology 56:
287–289, 2001 - Margolis RL: The Spinocerebellar Ataxias: Order Emerges from chaos. Curr Neurol and Neurosci Rep 2: 477–456, 2002
- Perlman SL: Spinocerebellar degeneration: an update. Curr Neurol Neurosci Rep 2: 331 – 341, 2002
Silveira I et al., Trinucleotide repeats in 202 families with ataxia. A small expanded (CAG)n allele at the
SCA17 locus. Arch Neurol 59: 623–629, 2002 - Bang OY et al., Clinical and Neuroradiological features of patients wIth spinocerebellar ataxias from Korean
kindreds. Arch Neurol 60: 1566–1574, 2003 - Bryer A et al., The hereditary adult‐onset ataxias in South Africa. J Neurol Sci 216: 47–54, 2003
- Bürk K et al., Cognitive deficits in spinocerebellar ataxia type 1, 2, and 3. J Neurol 250: 207–211, 2003
- Okamoto K et al., MR features of diseases involving bilateral middle cerebellar peduncles. Am J Neuroradiol
24: 1946–1954, 2003 - Brusco A et al., Molecular genetics of hereditary spinocerebellar ataxia. Mutation analysis of
spinocerebellar ataxia genes and CAG/CTG repeat expansion detection in 225 Italian families. Arch Neurol
61: 727–733, 2004 - De Michele G et al., A pathogenetic classification of hereditary ataxias: Is the time ripe? – J Neurol 251: 913–
922, 2004 - Xia H et al., RNAi suppresses polyglutamine‐induced neurodegeneration in a model of spinocerebellar
ataxia. Nat Med 10: 816–820, 2004 - van de Warrenburg BPC et al., Peripheral nerve involvement in spinocerebellar ataxias. Arch Neurol 61: 257–261, 2004
- Gatchel JR and Zoghbi HY: Diseases of unstable repeat expansion: mechanisms and common principles. Nat
Rev Genet 6: 743–755, 2005 - Gould VFC: Mouse models of Machado‐Joseph Disease and other polyglutamine spinocerebellar ataxias.
Am Soc Exp Neurother 2: 480–483, 2005 - Jiang H et al., Frequency analysis of autosomal dominant spinocerebellar ataxias in mainland Chinese
patients and clinical and molecular characterization of spinocerebellar ataxia type 6. Chin Med J 118(10):
837–843, 2005 - Juvonen V et al. The occurence of dominant spinocerebellar ataxias among 251 Finnish ataxia patients and
the role of predisponding large normal alleles in a genetically isolated population. Acta Neurol Scand 111:
154–1562, 2005 - Klockgether T: Ataxiekrankheiten. Diagnostisches Vorgehen und Therapie. Nervenarzt 20: 1275–1285,
2005 - Kraft S et al., Adult onset spinocrebellar ataxia in a Canadian movement disorders clinic. Can J Neurol Sci 32: 450–458, 2005
- Manto M‐U: The wide spectrum of spinocerebellar ataxias (SCAs). Cerebellum 4: 2–6, 2005
Tanaka M et al., A novel therapeutic strategy for polyglutamine diseases by stabilizing aggregation‐prone
proteins with small molecules. J Mol Med 83: 343–352, 2005 - Van de Warrenburg BP et al., Recent advances in hereditary spinocerebellar ataxias. J Neuropathol Exp
Neurol 64: 171–180, 2005 - Van de Warrenburg BPC et al., Age at onset variance analysis in spinocerebellar ataxias: a study in a Dutch‐
Frech cohort. Ann Neurol 57: 505–512, 2005 - Dragasevic NT et al., Frequency analysis and clinical characterization of different types of spinocerebellar
ataxia in Serbian patients. Mov Disord 21(2):187–191, 2006
- Rosenberg RN (Ed): Autosomal dominant cerebellar phenotypes: The genotype will settle the issue.
Järjestötoiminta
Potilasjärjestö: Neuroliitto ry; Ataksiaverkosto
Muita tietolähteitä
- The National Ataxia Foundatio: www.ataxia.org
- Sveriges socialstyrelsens kunskapsdatabas om ovanliga diagnoser innehåller information om fler än 300 ovanliga sjukdomar och tillstånd.