
Spinoserebellaariataksia tyyppi 13 (SCA13)
- ICD-10 numero G11.8, ORPHA: 98768, OMIM: 605259
Spinoserebellaari ataksia tyyppi 13 (SCA13) lukeutuu harvinaisten ataksiaa aiheuttavien sairauksien moninaiseen ryhmään. Ataksialla tarkoitetaan tahdonalaisten liikkeiden hallinnan ja koordinoinnin ongelmia. Ataksiasairauden taustalla on toimintahäiriö pikkuaivoissa tai niiden hermoratayhteyksissä. Ataksiaan johtavia syitä on lukuisia ja useimmiten ataksian taustalla onkin jokin varsin paljon tavallisempi sairaus kuin SCA13. SCA13 oli järjestyksessään kolmastoista tunnistettu virheellinen geenialue vallitsevasti periytyvien ataksiasairauksien joukossa. Toistaiseksi SCA13:n geenimutaation tunnistaminen ei ole onnistunut. Sairaus on kuvattu ranskalaisessa suvussa.
1890-luvulla kuvattiin ensimmäiset perheet, joissa etenevä pikkuaivoataksia todettiin peräkkäisissä sukupolvissa. Sairaus siis periytyi autosomissa dominantisti (AD) eli vallitsevasti. Sairautta esiintyi molemmilla sukupuolilla. 1900-luvulla eri puolilta maailmaa kuvattiin useita sukuja, joilla etenevä ataksiaoireisto todettiin peräkkäisissä sukupolvissa.
Jo vuonna 1863 oli lääketieteelliseen kirjallisuuteen kuvattu niin ikään pikkuaivojen ja selkäytimen toimintavaurioista johtuva perinnöllinen ataksiasairaus Friedreichin ataksia (FRDA), missä sairastumista todettiin vain sisarussarjassa, muttei vanhemmilla. Vanhemmat olivat sairauden oireettomia kantajia. FRDA periytyi autosomissa resessiivisesti (AR). Aina ei ataksiaa sarastavan suvusta muita samankaltaisesti oirehtivia perheenjäseniä löydetä.
Yrityksiä vallitsevasti periytyvien ataksiasairauksien (AD-ataksiat) ryhmittelyksi tehtiin useampiakin 1900-luvulla. 1980-luvun alkupuolella englantilainen neurologi Anita Harding pyrki yhtenäistämään varsin kirjavaksi muodostuneen nimikkeistön ja jakoi AD-ataksiat kliinisten oireiden perusteella kolmeen pääryhmään, joista hän käytti nimitystä ADCA-sairaudet tyypit I, II ja III. ADCA kirjainlyhenne on peräisin sanoista Autosomi, Dominantti, Cerebellum (pikkuaivot) ja Ataksia. ADCA tyyppi I:een lukeutuivat ne potilaat, joilla pikkuaivojen toimintahäiriöiden lisäksi todettiin merkkejä aivorungon tumakkeiden sekä selkäytimen toimintahäiriöistä. Osalla potilaista todettiin myös ääreishermoston vaurioita. ADCA tyyppi II:ssa edellä mainittujen oireiden lisäksi todettiin aina silmän verkkokalvon ja terävän näön keskuksen (makulan) rappeutuminen. ADCA tyyppi III:ssa oireet rajoittuivat pääasiallisesti pikkuaivojen toimintahäiriöihin.
ADCA tyyppi I ja III ovat osoittautuneet geneettisesti huomattavan moninaiseksi sairausryhmäksi. Virheellisen geenialueen tunnistaminen on johtanut käytäntöön nimittää sairautta geenin tunnistamisjärjestyksen mukaisesti SCA1-, SCA2-, SCA3-sairaudeksi jne. Tällä hetkellä tunnetaan yli kaksikymmentä SCA-sairauksien geenialuetta (SCA1-8, SCA10-22 sekä DRPLA). SCA13:n geenivirheen sijainti (kromosomi 19 q13.3 – q13.4) paikallistettiin vuonna 2000. Sairauteen liittyvät neurologisia oireita on raportoitu vain yhdestä ranskalaisesta perheestä. SCA13 geenimutaation tunnistaminen niin sanotulla suoralla DNA-testillä ei toistaiseksi ole mahdollista.
SCA13:n taudinkuva
SCA13:een liittyvät aina tasapainovaikeudet, kävelyn epävarmuus sekä raajojen koordinaatio-ongelmat, epätarkkuus ja hapuilu. SCA13:n oireet alkavat tavallisesti jo lapsuudessa. Taudinkuva vastaa AD-sairauksien kliinisen luokituksen ADCA tyyppi I:tä. Taudinkuvaan liittyvät ataksiaoireiden ohella liikunnallisen ja älyllisen kehityksen hidastuminen sekä puheen häiriöt (dysartria).
SCA13:n taudinkuvaan kuuluvat:
- tasapainon ja kävelyn epävarmuus (kävelyataksia)
- hienomotoriikan epätarkkuus
- hidastunut motorinen kehitys
- puheen sammaltaminen ja artikulaation epäselvyys (dysartria)
- lievä henkisten toimintojen taantuminen
- sairaus periytyy autosomissa dominantisti
Osalla todetaan:
- silmänvärve (nystagmus) ja muita silmien liikehäiriöitä
- lihasjänteyden nousu (spastisiteetti) ja vilkastuneet jänneheijasteet (hyperrefleksia)
- yhdellä tutkituista oli poissaolokohtauksia
Pitkään sairastaneilla voidaan todeta (> 50 vuotialla potilailla):
- nielemisen ongelmia (dysfagiaa)
- rakon pidätysvaikeutta (pakkoinkontinenssia)
- liikkeiden hitautta
- autonomisen hermoston toimintahäiriöitä
- tuntomuutoksia
SCA13 alkaa tasapainon ja motorisen kehityksen ongelmina, kömpelyytenä ja liikekankeutena usein jo lapsuudessa. Liikunnalliset taidot eivät välttämättä kehity samaan tahtiin kuin muilla samanikäisillä. Juokseminen, osallistuminen pallopeleihin, hiihtäminen osoittautuvat hankaliksi. Hienoinen tasapainohaitta voi haitata portaiden laskeutumista – kaidetuki on tarpeen ja tasapaino horjahtaa äkillisissä liikkeissä. Liikunnan ongelmien ilmaantuminen on varsin yksilöllistä. Jalkojen lihasjänteys on useimmilla kohonnut (spastisiteetti). Liikunnan apuvälineet saattavat olla tarpeen jo lapsuudessa.
Käsinäppäryys heikkenee. Pienten esineiden käsittely tuottaa ongelmia. Käsien osumatarkkuus heikkenee ja käsillä tekeminen vaatii aikaisempaa enemmän keskittymistä.
Sairauteen liittyy joko vaikea tai keskivaikea puheen sammallus. Joillakin todetaan myös nielemisen ongelmia. Silmien liikehäiriöistä tavanomainen on horisontaalinen nystagmus, mutta katseen kohdistamisvaikeutta samoin kuin silmien liikkeiden rajoittumista on todettu.
SCA13:een liittyy viivästymä henkisessä kehityksessä (mentaalinen retardaatio), joskin useimmilla henkisten toimintojen heikkeneminen on vain lievää tai korkeintaan keskivaikeaa. Niin ajan kuin paikankin tunnistaminen säilyvät. Vanhemmilla tutkituista ( 28 – > 50 vuoden ikäisillä) on todettu alentuneita pisteitä dementia-asteikolla (MMSE) suoritetuissa mittauksissa. MMSE-pisteet olivat 23–25 /30 vastaten lievää dementoitumista.
Yhdellä tutkituista todettiin lyhytkasvuisuus. Yhdellä leikki-ikäisenä oirehtineesta todettiin poissaolokohtauksia. Sairauteen saattaa liittyä myös autonomisen hermoston toimintahäiriöitä. Tihentynyttä virtsaamisen tarvetta todettiin, suoliston toimintamuutoksia ei ole kuvattu. Potilailla on todettu kiertävää pään pakkoliikettä (tortikollista).
SCA13:n oireet todetaan tavallisimmin jo lapsuudessa: motorinen kehitys hidastuu ja sairauteen liittyy myös oppimisen vaikeuksia. Yhdellä tutkituista sairaus alkoi vasta 45 vuoden iässä, nuorimmat sairastuneet ovat olleet alle vuoden ikäisiä. Pääsääntöisesti sairaus on hidaskulkuinen, mutta vuosien mittaan oirekirjo moninaistuu. Sairaus voi lyhentää elinikää, mutta vanhimmat tutkitut ovat olleet vanhuusikäisiä. Osalla lapsena sairastuneista henkiseen kehitykseen liittyvät ongelmat todetaan ennen motoristen taitojen heikkenemistä.
SCA13:n diagnoosi ja erotusdiagnoosi
Neurologisessa tutkimuksessa todetaan pikkuaivojen toimintahäiriöön sopivia löydöksiä: tasapainovaikeus, kävelyn ongelmat, sekä käsien että jalkojen osumatarkkuuden heikkeneminen. Hienomotoriset suoritukset ovat epätarkkoja ja vastakkaiset liikesuoritukset ovat epärytmisiä. Alaraajojen lihastonus on useimmilla lisääntynyt ja jänneheijasteet vilkastuneet. Lapsena sairastunut on kömpelö ja epävarma liikkuessa eikä motoristen ikävakioitujen testien läpivienti välttämättä onnistu. Henkiseen kehitykseen liittyvät ongelmat, samoin kuin puheen epäselvyys todetaan.
Aivojen magneettitutkimuksessa nähdään pikkuaivomadon (vermis) rappeuma, sen sijaan pikkuaivohemisfäärit ovat varsin hyvin säilyneet. Rappeutumaa nähdään myös aivorungossa. Alimmainen aivokammio (IV ventrikkeli) on suurentunut. Aivorungon herätepotentiaalitutkimuksissa ei todeta muutoksia. Katseensuuntainen silmänvärve (nystagmus) oli kaikilla dokumentoitavissa.
SCA13:n diagnoosia ei voida toistaiseksi määrittää suoralla geenitestillä ja näin ollen diagnoosimahdollisuutta epäiltäessä onkin harkittava yhteyden ottoa suoraan sairauden geenivirhettä tutkiviin laboratorioihin.
Erotusdiagnostiikassa tulevat kyseeseen ennen kaikkea muut SCA-sairaudet ja niistä erityisesti ne, joissa todetaan ADCA tyyppi I:n kaltainen oireisto. SCA13:een liittyvä henkisten toimintojen taantuminen erottaa sen monista muista SCA-sairauksista, samoin kuin oireiston tavanomainen alkaminen jo varhain lapsuudessa. Kuvatussa suuressa suvussa ei todettu periytymistä isältä pojalle.
SCA13:n esiintyminen
Toistaiseksi SCA13 on kuvattu vain ranskalaisessa suvussa, johon kuului useampia perheitä.
SCA13:n periytyminen
SCA13:een ei toistaiseksi ole käytettävissä suoraa sairauden diagnoosia varmentavaa geenitestiä. Sairaus periytyy autosomissa dominantisti . SCA13:a esiintyy yhtä suurella todennäköisyydellä molemmilla sukupuolilla ja sairastuneen vanhemman kaikilla lapsilla on 50 %:n todennäköisyys sairastumiseen.
Antisipaatiolla tarkoitetaan sairauden puhkeamista keskimäärin varhemmin seuraavassa sukupolvessa. Lähes kaikkiin tunnettuihin SCA-sairauksiin liittyy antisipaatio. Osasyynä tähän on monessa muussa SCA-sairaudessa todetun geenimutaation – CAG-jakson – taipumus pidentyä seuraavassa sukupolvessa. SCA13:n geenimutaatio ei tutkituissa suvuissa osoittautunut CAG-jakson pidentymäksi, eikä perheissä ole todettu myöskään viitteitä sairauden varhaistumisesta alenevissa sukupolvissa.
Perinnöllisyysneuvonnassa käynti on aiheellista järjestää kaikille perheenjäsenille. Neuvonnassa annetaan sairastuneelle ja hänen perheelleen tietoa niin sairauden laadusta kuin myös sen perinnöllisyydestä. Neuvonnassa annetaan myös tietoa ennakoivan geenitestin (prediktiivinen geenitesti) mahdollisuudesta.
SCA13 periytyy autosomissa dominantisti, mistä johtuen perheenjäsenen sairastumisen todennäköisyydestä voidaan sanoa seuraavaa (ks. Kuva 1)::
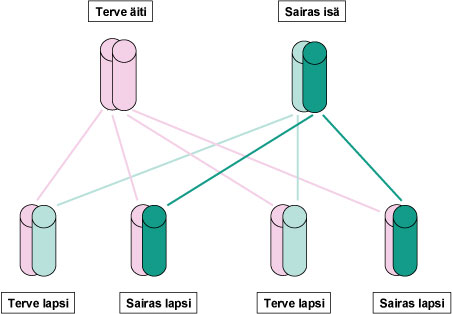
- Sairastuneen vanhemmat: Useimmilla niillä, joilla diagnostisoidaan SCA13, todetaan myös toisella vanhemmista kliininen sairaus.
- Sairastuneen sisarukset: Biologisilla sisaruksilla on 50 %:n todennäköisyys sairastumiseen.
- Sairastuneen omat lapset: Sairastuneen lapsilla on 50 %:n todennäköisyys sairastumiseen.
Ennakoivan geenitestin (prediktiivinen geenitesti) tekeminen ei toistaiseksi ole SCA13:ssa mahdollista. Myöskään sikiödiagnostiikka ei ole mahdollista.
SCA13:n hoito ja kuntoutus
Sairauden hoitoon ei ole käytettävissä sairautta parantavaa lääkehoitoa. Joitakin SCA13:een liittyviä oireita, kuten rakontoimintahäiriöitä tai epilepsiaa voidaan hoitaa käytössä olevin lääkkein. Vitamiinilisiä on suositeltu – erityisesti silloin, kun tavanomaisesta ravinnosta niitä ei todennäköisesti riittävästi kerry.
Perinnöllisyysneuvonta on oleellinen osa SCA13:n diagnostiikkaa ja hoitoa. Neuvontaa tarvitsevat sairastuneen lisäksi myös muut perheenjäsenet.
SCA13-sairauden aiheuttamat toimintarajoitteet etenevät yksilöllisesti, mutta oireiden alkaminen usein jo varhaislapsuudessa merkitsee kuntoutuksellisten toimien ajoittamista huomattavasti varhaisempaan vaiheeseen kuin useimmissa muissa SCA-sairauksissa. Kuntoutuksessa tulee huomioida niin lapsen liikunnallisen kehityksen tukeminen kuin sairauden mahdollisesti tuomat vaikeudet oppimisessa. Oleellista on, että tarvittavat kuntoutustoimet kartoitetaan sairauden tultua diagnosoiduksi.
Sairastunutta ja hänen perhettään tulee informoida sopeutumisvalmennuskursseista. Kursseilla annetaan tietoa sairaudesta ja kurssilla on mahdollisuus tavata muita sairastuneita.
Kuntoutustoimet kartoitetaan sairauden tultua diagnosoiduksi ja kuntoutustoimet kirjataan kuntoutussuunnitelmaan. Kuntoutussuunnitelman laatii hoidosta vastaava lääkäri. Suunnitelman ajantasaisuus on syytä tarkistaa kontrollikäyntien yhteydessä.
Yliopistosairaaloissa ja keskussairaaloissa on kuntoutusohjaaja. Terveyskeskuksissa toimivat kuntoutustyöryhmät. Nämä tahot huolehtivat suunniteltujen kuntoutustoimien järjestämisestä. Ataksiaa sairastavan kuntoutustoimissa moniammatillinen yhteistyö on perusteltua. SCA13-potilaan kuntoutuksessa arvioidaan niin fysioterapian ja toimintaterapian tarve. Puheterapeutin konsultaatio on tärkeää silloin, kun SCA13-potilaalla havaitaan puheeseen tai nielemiseen liittyviä ongelmia.
Sairastuminen etenevään sairauteen voi aiheuttaa muutoksia ammatinvalinnassa tai työelämässä. SCA13-sairauden oireita on usein todettavissa jo lapsuudessa. Sairauteen liittyy usein liikunnallisten ongelmien lisäksi myös oppimisen vaikeutta. Kuntoutustoimien tarkoituksena on auttaa sairastunutta selviytymään opiskeluissa sekä työ- ja arkielämässä huolimatta sairauden tuomista rajoitteista. Koulussa ja työelämässä jatkamista voidaan tukea monin eri tavoin. Tieto sairauden oireista ja sen luonteesta auttaa ratkaisuissa.
Liikunnallisten aktiviteettien tukeminen ja ylläpitäminen on arvokasta. Fysioterapian tarve arvioidaan kuntoutussuunnitelmaa laadittaessa. Fysioterapeutti on liikeharjoitusten asiantuntija ja samalla kannustaa ja opastaa omatoimisessa kunnon ylläpidossa. Terapeutti kykenee hyvin seuraamaan sairauden aiheuttamien liikuntahaittojen laatua ja mahdollista liikunnan apuvälineiden tarvetta. Kodin turvallisuutta lisäävät ja huonon tasapainon edellyttämät asunnonmuutostyötarpeet välittyvät mutkattomasti fysioterapeutin tai toimintaterapeutin kautta kotikunnan asianomaisille viranomaisille.
Puheterapia sekä kommunikaatiota helpottavat apuvälineet voivat tulla ajankohtaiseksi sairauden edetessä. Säännöllisen puheterapian tarve ei useinkaan ole tarpeen, mutta puheterapeutin käynnillä saadut ohjeet voivat merkittävästi selkeyttää ilmaisua. Nielemisen ongelmat kuuluvat SCA13:ssa vasta sairastumisen myöhäisvuosiin.
SCA13:n perustutkimus
Neuropatologisia tutkimusraportteja ei toistaiseksi ole käytettävissä.
SCA13:n geenimutaatio on kromosomissa 19 alueella q13.3 – 13.4. Kyseiselle geenialueelle on aikaisemmastaan paikallistettu useiden tunnettujen proteiinien geenejä: Bcl-2 assosioituvan X proteiinin (BAX) geeni, fosfolipaasi A2:n geeni ja kalmoduliinin geeni. SCA13-geeni voi määrittää todennäköisesti proteiinirakennetta jossakin näistä edellä mainituista proteiineista. Geenimutaation tarkka tunnistaminen ja sen ohjaaman proteiinin identifioiminen ovatkin ensisijaisia SCA13:n perustutkimuksen kohteita.
Ohjeita arkeen
- Hae tietoa sairaudesta – käytä hyväksesi alla lueteltuja tietolähteitä
- Älä jää yksin – vertaistuki on usein korvaamaton tuki
- Hakeudu sopeutumisvalmennuskurssille, ota yhteyttä potilasyhdistykseen, kysy tukihenkilöistä
- Sovella terapeuteiltasi saamiasi ohjeita arkipäivän toimissasi – tasapainoa ja liikeratoja on mahdollista harjoittaa
- Keskity kulloiseenkin tekemiseesi: ataksiaa sairastavan syöminen ja puhuminen eivät välttämättä onnistu yhtäaikaisesti, myös käveleminen ja puhuminen yhtäaikaisesti saattaa olla ongelmallista
- Älä arastele sairautesi asettamia rajoitteita tai mahdollista avuntarvettasi
- Lyhyt esite tai ajokortin tapainen invalidikortti helpottaa tilanteissa, joissa tasapainovaikeuksien ja liikehäiriöiden syyksi epäillään juopumustilaa – pyydä se joko hoitavalta lääkäriltä tai sopeutumisvalmennuskurssin lääkäriltä
- Muista, että sairastuminen ei ole sinun tai läheistesikään syytä
- Säilytä aikaisemmat aktiviteettisi siinä määrin kuin mahdollista ja hae uusia aktiviteetteja mahdollisten menetettyjen tilalle
- Ole aktiivisesti mukana laatimassa kuntoutus- tai palvelusuunnitelmasi tavoitteita ja sisältöä – tavanomaisen fysioterapian ohella on myös muita toiminnallisuutta ylläpitäviä ja kohentavia terapiamuotoja
- Ota yhteyttä Neuroliittoon; saat tietoa niin ataksiaverkon toiminnasta kuin myös tukihenkilöistä
Sopeutumisvalmennus- ja kuntoutuspalvelut
Perinnölliset ataksiasairaudet ovat harvinaisia sairauksia kaikkialla maailmassa. Suomessa Maskun neurologinen kuntoutuskeskus on valtakunnallinen neurologisten sairauksien resurssikeskus. Tietoa Maskun neurologisen kuntoutuskeskuksen kursseista.
Kirjoittaja
Riitta Rinne, LL, neurologian erikoislääkäri, Maskun neurologinen kuntoutuskeskus
Julkaistu 19.12.2002
- Kirjallisuusviitteet
- Rosenberg RN (Ed): Autosomal dominant cerebellar phenotypes: The genotype will settle the issue
Neurology 40: 1329–1331, 1990 - Harding AE: Ataxic disorders. Kirjassa: Neurology in clinical practice, 337–346. Toim. WG Bradley, RB
Daroff, GM Fenichel and CD Mardsen. Butterworth‐Heinemann, Boston 1991(a) - Harding AE: Cerebellar and spinocerebellar disorders. Kirjassa: Neurology in clinical practice, 1603–1623.
Toim. WG Bradley, RB Daroff, GM Fenichel and CD Mardsen. Butterworth‐Heinemann, Boston 1991(b) - Rosenberg RN (Ed): Autosomal dominant cerebellar phenotypes: The genotype has settled issue. Neurology
45: 1–5, 1995 - Rosenberg RN (Ed): Spinocerebellar ataxias and ataxins. New England J Med 333 (20): 1351–1352, 1995
Grewal RP et al.: Clinical and genetic analysis of a distinct autosomal dominant spinocerebellar ataxia.
Neurology 51: 1423–1426, 1998 - Klockgether T and Evert B: Genes involved in hereditary ataxias. Trends Neurosci 21: 413–418, 1998
- Subramony SH and Nance M: Diagnosis and Management of the inherited ataxias. The Neurologist 4: 327–338, 1998
- Ashizawa T (Ed): Repeats may not be everything in anticipation. Neurology 53: 1164–1165, 1999
Herman‐Bert A et al: Mapping of the spinocerebellar ataxia 13 to chromosome 19q13.3‐q13.4 ia a family
with autosomal dominant cerebellar ataxia and mental retardation. Am J Hum Genet 67: 229‐235, 2000 - Subramony SH and Filla A (Ed): Autosomal dominant spinocerebellar ataxias ad infinitum? Neurology 56:
287–289, 2001 - Tan E‐K and Ashizawa T: Genetic Testing in Spinocerebellar Ataxias. Defining a Clinical Role. Arch Neurol 58:
191–195, 2001 - Perlman SL: Spinocerebellar Degenerations: An Update. Curr Neurol and Neurosci Rep 2: 331–341, 2002
- Margolis RL: The Spinocerebellar Ataxias: Order Emerges from Chaos. Curr Neurol and Neurosci Rep 2: 477–456, 2002
- Rosenberg RN (Ed): Autosomal dominant cerebellar phenotypes: The genotype will settle the issue
Järjestötoiminta
Potilasjärjestö: Neuroliitto ry; ataksiaverkosto
Muita tietolähteitä
- The National Ataxia Foundation: www.ataxia.org
- Sveriges socialstyrelsens kunskapsdatabas om ovanliga diagnoser innehåller information om fler än 300 ovanliga sjukdomar och tillstånd.