
Spinoserebellaarinen ataksia tyyppi 3 (SCA3)
- ICD-10: G11.8 Muut määritetyt perinnölliset ataksiat, OMIM: 109150, ORPHA: 98757
- Muut sairaudesta käytetyt nimet:
- Machado-Josephin tauti (MJD)
- Spinoserebellaarinen atrofia III
- Azorian neurologinen sairaus
- Spinopontinen atrofia
- Nigrospinodentaalinen degeneraatio
Spinoserebellaarinen ataksia tyyppi 3 (SCA3) on pikkuaivojen ja niiden hermoratayhteyksien toimintahäiriöihin johtava etenevä sairaus. SCA3 periytyy autosomisesti vallitsevasti eli dominantisti (AD). Sairaudesta usein käytetty nimi on Machado-Josephin tauti. Oireiden alkaminen ja niiden eteneminen vaihtelevat sairastuneilla merkittävästi ja oirekuva on yhteydessä sairauden alkamisikään (SCA3 tyypit 1–3). Pikkuaivo-oireiden (tasapainovaikeus, hienomotoriikan heikkeneminen ja puheen sammallus) lisäksi SCA3:ssa todetaan lihasjänteyden lisääntymistä (rigiditeetti, spastisiteetti), pakkoliikkeitä ja silmien liikehäiriöitä sekä rajoittumista. Ääreishermojen rappeutuminen (polyneuropatia) sekä kognitiivinen heikkeneminen liittyvät SCA3:n oirekuvaan. Sairautta aiheuttava geenimutaatio tunnetaan ja sen määrittäminen verinäytteestä varmistaa SCA3-diagnoosin. SCA3 lukeutuu polyglutamiiniataksioihin (PolyQ-ataksiat).
SCA3 alkaa tavallisimmin 40 vuoden iässä; keskimäärin 41,5 ± 2,3 vuoden iässä. Sairauden kestossa on eri aineistoissa jossain määrin poikkeavia havaintoja, tavallisimmin se on 10,5 ± 1,3 vuotta (1–20 vuotta), mutta on potilaita, joiden sairaus etenee hitaammin ja sairastamisaika on 20 vuotta. Nuorimmat SCA3 diagnoosin saaneet ovat olleet alle 10 vuoden ikäisiä ja vanhimmat 70 vuoden ikäisiä. SCA3 on monioireinen sairaus. Sairauden edetessä todetaan kaikilla tasapainoheikkous ja puheen kangertaminen. Oireiden alkamisiällä on vaikutusta oirekuvaan, josta syystä SCA3 ryhmitellään kolmeen alatyppiin (Taulu 1.).
SCA3:n oireet vaihtelevat sairastumisiästä riippuen
SCA3-tyyppi I
- Alkaa alle 20 vuoden iässä (jo lapsuudessa)
- Tyypillistä oireissa: Lihastonuksen nousu (rigiditeetti), liikkeiden hitaus (bradykinesia), pakkoliikkeet (dystonia ja korea)
SCA3-tyyppi II
- Alkaa 20–50 vuoden iässä
- Tyypillisiä oireita: tasapainovaikeus, ataksia, lihastonuksen nousu (spastisiteetti), pakkoliikkeet (dystonia, korea)
SCA3-tyyppi III
- Hieman alle 50 vuoden iässä tai myöhemmin
- Tyypillisiä oireita: tasapainovaikeus, ataksia, senso-motorinen polyneuropatia, voimien heikkeneminen ja lihaskato
Perheenjäsenten sairastumisiässä, samoin kuin taudinkulussa poikkeuksellisen runsasta vaihtelua. Kaikissa suvuissa ei yllä olevan kaltaista oireiden alkamisiän perusteella määräytyvää taudinkuvaa ole todettu, vaan taudinkuva kaikilla sairastuneilla muistuttaa SCA3 tyyppi II:a (Kuva 1.)
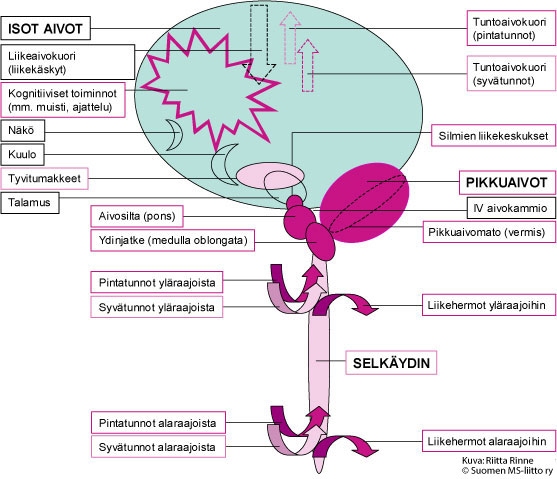
Kuva 1. Aikuisiällä alkavan SCA3:n aiheuttama neurologinen oireisto johtuu pikkuaivojen, aivorungon sekä tyvitumakkeiden ja niiden hermoratayhteyksien toiminnan heikkenemisestä. Kuvassa on tummalla violetilla merkitty hermoston alueet, joissa SCA3-sairaus aiheuttaa aina muutoksia ja vaaleammalla violetilla ne hermoalueet, joissa muutoksia voi esiintyä.
Nuorena, tavallisesti ennen 20 ikävuotta alkava SCA3-oireisto poikkeaa aikuisiällä alkavasta sairaudesta. Tyypillistä on liikkeiden kankeus ja hitaus (bradykinesia) sekä pakkoliikkeet (dystonia ja korea). Oireiden eteneminen on yleensä nopeampaa kuin myöhemmin alkaneessa sairaudessa. Sairauden edetessä todetaan tasapainohäiriöitä sekä liikkeiden hallintavaikeuksia. Puhe- ja nielemisongelmat kuuluvat taudinkuvaan sekä silmien liikehäiriöt.
Aikuisiän SCA3-sairauden taudinkuvaan kuuluvat:
- tasapainon ja kävelyn epävarmuus (kävelyataksia)
- vartalon ja raajojen liikkeiden hallintavaikeus (ataksia)
- alaraajojen lihasjänteyden lisääntyminen (spastisiteetti, rigiditeetti) ja kiihtyneet jänneheijasteet (hyperrefleksia)
- silmänvärve (nystagmus) ja kaksoiskuvia
- silmien liikehäiriöt ja silmien liikerajoitukset; katseen suuntaaminen haluttuun katsesuuntaan ei onnistu
- puherytmin hidastuminen ja artikulaation epäselvyys (dysartria)
- suun ja nielun alueen lihasheikkous; suun ympärillä todettaan lihasnykäyksiä (faskikulaatiota)
- kielen kuihtuminen (kieliatrofia)
- nielemisvaikeudet (dysfagia)
Osalla SCA3-potilaista todetaan:
- pakkoliikkeitä (dystoniaa sekä koreaa) todetaan erityisesti nuorena sairastuneilla, mutta osalla myöhemminkin sairastuneista
- lihasten surkastuminen (amyotrofia) ja jänneheijasteiden sammuminen (arfleksia)
- tuntojen heikkeneminen raajojen kärkiosissa ja vibraatiotunnon häviäminen
- yläraajojen vapinaa
- autonomisen hermoston säätelyhäiriöitä
- keskittymiseen sekä toimintojen suunnitteluun liittyviä ongelmia
Aikuisiällä alkavan SCA3:n taudinkuva ja oireiden eteneminen
Aikuisiällä alkavassa SCA3:ssa tasapainon epävarmuus ja kävelyn horjahtelu (kävelyataksia) ovat tavalliset ensioireet. Sairauden edetessä tasapainon ylläpitäminen seisoessakin on vaikeaa. SCA3:ssä todetaan myös selvä vartalon hallintavaikeus: istuma-asento horjahtaa herkästi ja kurkottelut ovat hankalia. Hienomotoriikka hidastuu ja kömpelöityy. Käsien osumatarkkuus heikkenee. Monimutkaiset liikesuoritukset käyvät mahdottomiksi eikä uusia liiketaitoja opita.
Sairauden ensivuosina lihasten tonus on etenkin alaraajoissa kohonnut (spastisiteetti) ja heijasteet vilkastuneet, mutta sairauden edetessä lihasvoima heikkenee ja lihakset kuihtuvat (amyotrofiaa). Sairauden edetessä myös jänneheijasteet vaimenevat ja sammuvat. Lihasheikkoutta ja lihasten surkastumista todetaan niin kasvoissa kuin raajoissakin.
Puheen epäselvyys ja artikulaation ongelmat kuuluvat kaikilla oirekuvaan. Kasvolihasten heikkous ja surkastuminen kuuluvat oirekuvaan. Suun ympärillä todetaan hienoisia lihasnykinöitä (faskikulaatioita) sekä lihasvärinöitä (fibrillatiota) ja lihaskatoa (atrofiaa) kielessä. Nielemisvaikeudet ovat potilailla tavallisia. Sairauden edetessä nielmisheijaste sammuu ja yskiminen heikkenee aiheuttaen aspiraatioriskin.
SCA3:een liittyvät silmien liikehäiriöt. Silmänvärvettä (nystagmusta), katseensuuntainen nystagmus sekä kaksoiskuvia todetaan erityisesti sairauden ensi vuosien aikana. Katseen kohdistaminen rajoittuu vähittäisesti ja seurauksena on pään kääntöliike katsottaessa. Silmien liikerajoituksista ensimmäisenä todetaan ylöspäin katsomisen vaikeus. Sairauden edetessä silmien liikkeet rajoittuvat siinä määrin, että katse muuttuu ”tuijottavaksi”. Näön heikkenemistä ei ole todettu.
Osalla todetaan pakkoliikkeitä, dystoniaa ja koreaa. Dystonia on raajojen kiertävää, vetävää pakkoliikettä tai pään tahatonta kiertymistä. Korea on epätarkoituksellista ja epärytmistä liikehdintää raajoissa. Nivelten asentotunto ja värinätunto heikkenevät. Kosketus- ja kiputunto voivat alentua. Lihaskramppeja ja yöunta häiritsevää jalkojen ”levottomuutta” on joka toisella.
Niin rakon- kuin suolentoiminnan häiriötä on raportoitu vaihtelevasti. Autonomisen hermoston toimintahäiriöt liittyvät oirekuvaan ja samanaikaisesti saattaa esiintyä useita oireita. Tavallisia ovat kylmänarkuus, ortostaattinen hypotonia, silmien ja suun kuivuminen sekä lähelle katsomisen vaikeus. Yöuni saattaa olla katkeilevaa alaraajaoireiden takia (levottomat jalat) ja päiväväsymystä esiintyy. Potilailla on todettu muitakin unihäiriöitä: nukahtamisvaikeutta on joka toisella ja toistuvia painajaisunia näkee joka kymmenes.
Lapsuudessa alkavaan sairauteen (SCA3 tyyppi I) on kuvattu liittyvän laaja-alaista henkisten toimintojen taantumista, dementoitumista. Aikuisiällä alkavaan sairauteen voi liittyä ongelmia toimintojen suunnittelussa sekä keskittymiseen ja tarkkaavaisuuteen liittyviä häiriöitä. Kielellisessä ja visuaalisessa muistissa on todettu heikkenemistä, samoin sanasujuvuudessa. Heikkenemistä on todettu myös ympäristön havainnoinnissa sekä toimintojen sujuvuudessa. Aikuisiällä alkaneeseen SCA3:een on todettu harvoin liittyvän laaja-alaisia muistin ja toimintojen säätelyn ongelmia (dementiaa).
SCA3/MJD lyhentää elinikää. Naisella sairaus etenee suhteellisesti nopeammin kuin vastaavan ikäisenä sairastuneella miehellä. Toiminnallisen haitan nopeampi kertyminen ei kuitenkaan heijastu naisten varhaisempana kuolleisuutena. Samassakin perheessä sisarusten sairastumisikä vaihtelee huomattavasti, samoin kuin oireiden eteneminen ja niiden moninaisuus.
SCA3:n periytyminen
SCA3 periytyy autosomisesti vallitsevasti eli autosomissa dominantisti (AD). Sairautta aiheuttava geenivirhe tunnistettiin kromosomissa 14(q24.3-q31) vuonna 1994. Geenitesti varmistaa SCA3:n diagnoosin. SCA3-sairauden geeni on ATXN3/MJD-geeni ja sen tuottama valkuaisaine on Ataksiini – 3/MJD1. SCA3-sairaudessa geenimutaatio on geenin luentakehyksen sisäinen ylipitkä, epävakaa CAG-jakso (dynaaminen mutaatio). Normaalissa ATXN3-geenissä on < 45 (tavallisimmin 12–36) CAG-jaksoa. SCA3-sairauden geenissä on yli 44 CAG-jaksoa (tavallisimmin 53–86 CAG-jaksoa). CAG-nukleotidikomikko ohjaa aminohappo glutamiinin syntymisen. Seurauksena on ylipitkä glutamiiniketju (PolyQ-ketju) Ataksiini-3:ssa (Kuva2.)
Ylipitkän CAG-jakson dynaamisuus merkitsee CAG-jakson taipumusta pidentyä seuraavassa sukupolvessa. CAG-jakson pidentymistaipumus on havaittu etenkin sairauden periytyessä isältä. CAG-jakson pituuden ja oireiden puhkeamisen välillä on todettu käänteistä korrelaatiota: pitkä CAG-jakso on yhteydessä oireiden varhaiseen puhkeamiseen ja nopeaan taudinkulkuun (genotyyppi – fenotyyppi korrelaatio). Sairauden varhaistumista seuraavassa sukupolvessa kutsutaan antisipaatioksi. SCA3:n geenimutaatiossa CAG-jakso on pidentynyt.
SCA3:n fenotyyppi-genotyyppi korrelaatioita
SCA3 tyyppi I:ssä eli lapsuudessa sairastuneilla CAG-jakson pituus on aina > 75, tavallisimmin > 80 CAG-jaksoa. CAG-jakson pidentyminen 54–56:een voi aiheuttaa sairauden, mutta ei kaikilla SCA3-suvun jäsenillä. SCA3-sairaudessa tietynpituisilla CAG-mutaatioilla (genotyyppi) on todettu olevan merkitystä sairauden oirekuvaan myös aikuisiällä alkavassa SCA3-sairaudessa (fenotyyppi). SCA3-CAG-54:ssä on ataksiaan liittyy vaikea ääreishermostovaurio ja SCA3-CAG-56:ssa autonomisen hermoston toimintahäiriöt hallitsevat oirekuvaa ataksian ohella.
CAG-jakson pituuden perusteella ei oireiden puhkeamisikää tai niiden etenemistä voi ennustaa. On todettu, että sisarussarjassa samanpituisen CAG-jakson omaavat voivat sairastua eri-ikäisinä, eivätkä sisarusten oireet etene samaan tahtiin. CAG-jakson pituus ei siis yksinomaisesti määrää SCA3-sairauden oireiden etenemistä.
DNA-testi varmistaa SCA3:n diagnoosin. DNA-testiä voidaan käyttää myös sairautta ennakoivana (prediktiivinen geenitesti). Määritys voidaan tehdä halutessa täysi-ikäisiltä oireettomilta, riskissä olevilta sukulaisilta tai sikiönäytteestä.
SCA3:n diagnoosi ja erotusdiagnoosi
SCA3-diagnoosiin ja erotusdiagnoosiin tarvitaan neurologinen tutkimus sekä sen perusteella arvioitavat laboratorio-, kuvantamis- ja muut hermotoimintojen tutkimukset. Muiden erikoisalojen konsultaatiot ovat usein perusteltuja. Perinnöllisyyslääkärin konsultaatio ja neuvonta sairastuneelle ja hänen sukulaisille on aina tarpeen.
Laboratoriotutkimukset verinäytteestä, lukuun ottamatta diagnostista DNA-testiä, ovat normaalit, samoin selkäydinnestenäyte (likvor-tutkimus). Magneettitutkimuksessa todetaan hermokudoskatoa pikkuaivomadossa (vermiksessä), aivorungossa pons-tumakkeissa (erityisesti ponsin yläosassa), tyvitumakkeissa etenkin substantia nigrassa, keskimmäisissä pikkuaivoreisissä, selkäydinjatkeessa (medulla oblongatassa) ja kaulaytimessä. IV aivokammio on laajentunut. Pikkuaivokupoleissa (hemisfääreissä) ei surkastumaa nähdä. ENMG-tutkimuksessa todetaan useimmilla ääreishermojen aksonaalinen vaurio tunto- ja liikehermoissa. Neuro-oftalmologinen tutkimus todentaa silmien liikehäiriöiden laadun ja auttaa niin sairauden diagnostiikassa kuin erotusdiagnostiikassa. Aivojen aineenvaihduntaa kuvaavassa positroni emissio tomografiassa (PET-tutkimus) on todettu alentunutta sokeriaineenvaihduntaa pikkuaivoissa, aivorungossa, talamuksessa ja putamentumakkeissa. Striatum-tumakkeissa todettiin alentunut dopamiiniaktiivisuus.
Erotusdiagnoosissa tärkein ryhmä on muut PolyQ-ataksiat, erityisesti SCA1, SCA2 ja SCA17. Periaatteessa myös muut SCA-sairaudet, joiden oireet eivät rajoitu puhtaasti pikkuaivo-oireisiin (ADCA I), tulevat kyseeseen. Muita erotusdiagnostiikassa huomioitavia sairauksia ovat mm. Huntingtonin tauti, perinnöllinen Parkinson tauti, perinnölliset polyneuropatiat (HSMN-sairaudet), MS-tauti ja aivojen laaja-alainen surkastuma eli MSA.
SCA3-diagnoosi on erittäin harvoin todettu myös ataksiapotilailla, joiden suvussa ei vastaavasti sairastuneita ole aikaisemmastaan diagnosoitu.
SCA3:n esiintyminen
Suomessa ei toistaiseksi SCA3-sairautta ole diagnosoitu. Machado-Josephin taudin nimellä tunnettu kliininen oireisto kuvattiin alun perin USA:ssa. Geeni oli tullut maahan portugalilaisten maailmanmatkaajien mukana 1600-luvulla. 1990-luvun puolivälissä osoitettiin Machado-Josephin taudin geenimutaatio identtiseksi Euroopassa tunnistetun SCA3-geenimutaation kanssa. SCA3-sukuja on USA:ssa, Kanadassa, Portugalissa, Intiassa, Japanissa, Brasiliassa. Keski-Euroopassa SCA3 on tavallisin nyt tunnetuista SCA-sairauksista ja Portugalissa tietyillä alueilla SCA3:n esiintyvyys on jopa 100:100 000. USA:ssa SCA3-geenimutaatio todetaan lähes 10 %:lla SCA-sairauksista.
SCA3:n aiheuttamat hermovauriot
SCA3:n neuropatologiset muutokset paikallistuvat spinoserebellaarisiin ratoihin, globus pallidusten ja subtalamisten tumakkeiden alueille. Aivohermojen III, IV, V, VI, VII, VIII tumakkeissa nähdään solukatoa, samoin pikkuaivojen dentatus-tumakkeissa. Selkäytimessä rappeutumaa on etusarvien soluissa ja Clarken pylväikössä. Ääreishermojen rappeutumaa todetaan vaihtelevasti. Solutuhoa ei ole pikkuaivojen hemisfääreissä tai alemmissa oliva-tumakkeissa poiketen valtaosasta muiden SCA-sairauksien löydöksistä. Myös caudatum- ja putamen-tumakeet säilyvät. Sen sijaan pikkuaivojen Purkinjen solut kuin myös aivorungon oliva-tumakkeet ovat kohtuullisen hyvin säilyneitä.
SCA3:n hoito ja kuntoutus
Ataksian lievittäminen käytössä olevin lääkkein ei onnistu. Vapinaoireeseen voi yrittää beeta-salpaajaa. Lisääntynyttä lihastonusta vähentävät spastisiteetin hoitoon tarkoitetut lääkkeet. Parkinsonin taudin lääkehoidossa käytettävät L-dopamiini tai dopamiinin vaikutusta tehostavat valmisteet (dopamiini-agonistit) auttavat usein levottomiin jalkoihin. Rakon toimintahäiriöihin on niin ikään saatavissa apua. Aloitettavan oirelääkkeen tehon arvioinnissa on oltava kriittinen. Etenevässä sairaudessa lääkkeen tarpeellisuutta ja sen annosta on syytä tarkistaa. Hoitava lääkäri antaa ohjeet annosmäärän säätelyyn. Pitkään käytössä olleiden keskushermostoon vaikuttavien lääkkeiden lopettamista ei saa tehdä äkillisesti.
Kuntoutustarpeen arvioiminen ja kirjaaminen kuntoutussuunnitelman ja seuranta ovat keskeinen osa SCA3-sairauden hyvää hoitoa. Kuntoutussuunnitelmaan kirjataan eri terapioiden tarve, tarvittavat apuvälineet ja muut selviytymistä tukevat keinot. Tarvittavat palvelut kirjataan palvelusuunnitelmaan.
Liikuntakyvyn säilymistä tuetaan fysioterapialla. Tasapainon, lihasten hallinnan sekä koordinaation harjoittaminen omaehtoisesti kannattaa. Soveltuvista liikunnan muodoista saa ohjeita fysioterapeutilta, samoin kuin opastusta lihashuollosta ja rentoutuksesta. Puheterapeutin arviot ja ohjeet ylläpitävät kommunikaatiotaitoja.
SCA3:een liittyy nielemisongelmia. Ruoka menee helposti ”väärään kurkkuun” eli henkitorveen (aspiraatio). Aspiraatio altistaa toistuville keuhkotulehduksille ja pahimmillaan tukehtumiselle. Aspiraation riski tutkitaan videofluorometrialla. Nielemisen turvallisuuteen voidaan vaikuttaa. Nielemisen koordinointiongelmissa on vältettävä karkeusasteeltaan vaihtelevia ruokia. Kiinteä ruoka ja nesteet on nautittava erikseen. Nielemisen turvallisuutta lisäävät ruuan ja nesteiden tasalaatuisuus ja ruokailun rauhoittaminen. Vatsanpeitteiden läpi asetettavaa PEG-letkua suositellaan silloin, jos ruokailu aspiraatiovaaran takia ei ole turvallista.
Niin kommunikaatiota, arkipäivän toimien suorittamista kuin myös liikkumista on mahdollista helpottaa apuvälinein. Sairauden aiheuttaessa lisääntyvää avun- ja hoivan tarvetta on tehtävä palvelusuunnitelma. Kuntoutussuunnitelman tavoin myös palvelusuunnitelman ajantasaisuus on sovitusti tarkistettava.
SCA3:n perustutkimuksesta
SCA3-sairauden tutkimukseen on kehitetty niin kärpäs- kuin hiirimalleja. Ataksiini-3/MDJ1-proteiinia esiintyy laajalti elimistön eri kudoksissa. Se paikallistuu soluissa pääsääntöisesti solulimaan. Patologisen Ataksiini-3/MJD1:n saostumista (aggregaatiota) niin solulimaan kuin tumaan on todettu. Erityisesti hermosolujen tumaan kertyvien Ataksiini-3-proteiinisaostumien ajatellaan liittyvän sairauden patogeneesiin. Tumaan kertyviä saostumia on todettu runsaasti aivorungon hermosoluissa, tyvitumakkeiden substantia niagrassa, globus pallidum-tumakkeissa sekä pikkuaivojen dentatus-tumakkeissa.
Kirjoittaja
Riitta Rinne, LL, neurologian erikoislääkäri, Maskun neurologinen kuntoutuskeskus
Julkaistu 14.10.2002. Päivitetty 16.6.2006
- Kirjallisuusviitteet
- Nakano KK et al: Machado disease: a hereditary ataxia in Portugenese immigrants to
Massachusettes. Neurology 22: 49 – 55, 1972 - Burt T et al., A dominant hereditary ataxia resembling Machado‐Joseph disease in Arnhem Land,
Australia. Neurology 43: 1750 – 1752, 1993 - Takiyama Y et al., The gene for Machado‐Joseph disease maps to human chromosome 14q. Nat
Genet 4: 300 – 304, 1993 - George‐Hyslop PS et al., Machado‐Joseph disease in pedigrees of Azorean descent is linked to
chromosome 14. Am J Hum Genet 55: 120 – 125, 1994 - Kawaguchi Y et al., CAG expansions in a novel gene for Machado‐Joseph disease at chromosome
14 q 32.1. Nat Genet 8: 213 – 215, 1994 - Maciel P et al., Correlation between CAG repeat length and clinical features in Machado‐Joseph
disease. Am J Hum Genet 57: 54 – 61, 1995 - Schöls L et al., Trinucleotide expansion within the MJD1 gene presents clinically as spinocerebellar
ataxia and occurs most frequently in German SCA patients. Hum Mol Genet 4: 1001 – 1005, 1995 - Stevanin J et al., Linkage disequilibrium at the Machado‐Joseph disease/spinal cerebellar ataxia 3
locus: evidence for a common founder effect in French and Portugenese‐Brazilian families as well
as a second ancesteral Portugene‐Azorean mutation. Am J Hum Genet 57: 1247 – 1250, 1995 - Twist EC et al., Machado‐Joseph disease maps to the same region of chromosome 14 as the
spinocerebellar ataxia type 3 locus. J Med Genet 32: 25 – 31, 1995 - Bürk K et al., Autosomal dominant cerebellar ataxia type I. Clinical features and MRI in families
with SCA1, SCA2 and SCA3. Brain 119: 1497 – 1505, 1996 - Dürr A et al., Spinocerebellar ataxia 3 and Machado‐Joseph disease: clinical, molecular, and
neuropathological features. Ann Neurol 39: 490 – 499, 1996 - Higgins JJ et al., Mutations in American families with spinocerebellar ataxia (SCA) type3: SCA3 is
allelic to Machado‐Joseph disease. Neurology 46: 208 – 213, 1996 - Klockgether T et al., Repeat length and disease progression in spinocerebellar ataxia type 3. Lancet
348: 830, 1996 - Maruff P et al., Cognitive deficits in Machado‐Joseph disease. Ann Neurol 40: 421 – 427, 1996
- Kaakkola S ja Rinne R: Ataksiat ja niiden erotusdiagnostiikka (katsaus). Duodecim 113: 1773 –
1782, 1997 - Grewal RP et al., Clinical and genetic analysis of a distinct autosomal dominant spinocerebellar
ataxia. Neurology 51: 1423 – 1426, 1998 - Klockgether T et al., The natural history of degenerative ataxia: a retrospective study in 466
patients. Brain 121: 589 – 600, 1998 - Lima M et al., Causes of death in Machado‐Joseph disease. A case‐control Study in the Azores
(Portugal). Arch Neurol 55: 1341 – 1344, 1998 - Moseley ML et al., Incidence of dominant spinocerebellar and Friedreich triplet repeats among
361 ataxia families. Neurology 51: 1666 – 1671, 1998 - Murata Y et al., Characteristic magnetic resonance imaging findings in Machado‐Joseph disease.
Arch Neurol 55: 33 – 37, 1998 - Onodera O et al., Progressive atrophy of cerebellum and brainstem as a function of age and the
size of the expanded CAG repeats in the MJD1 gene in Machado‐Joseph disease. Ann Neurol 43:
288 – 296, 1998 - Schmidt T et al., An isoform of ataxin‐3‐accumulates in the nucleus of neuronal cells in affected
brain regions of SCA3 patients. Brain Pathol 8: 669 – 679, 1998 - Schöls L et al., Sleep disturbance in spinocerebellar ataxias. Is the SCA3 mutation a cause of
restless legs syndrome? Neurology 51: 1603 – 1607, 1998 - Warrick JM et al., Suppression of a polyglutamine‐mediated neurodegeneration in Dorsophilia by
the molecular chaperone HSP70. Nature Genetics 23: 425 – 428, 1999 - Schöls L et al., Genetic background of apparently idiopathic sporadic cerebellar ataxia. Hum Genet
107: 132 – 137, 2000 - Storey E et al., Frequency of spinocerebellar ataxia types 1, 2, 3, 6, and 7 in Australian patients
with spinocerebellar ataxia. Am J Med Genet 95: 351‐357, 2000 - Tang B et al.: Frequency of SCA1, SCA2, SCA3/MJD, SCA6, SCA7, and DRPLA CAG trinucleotide
repeat expansions in patients with hereditary spinocerebellar ataxia from Chinese kindreds. Arch
Neurol 57: 540 – 544, 2000 - Gwinn‐Hardy K et al., Spinocerebellar ataxia type 3 phenotypically resembling Parkinson disease in
a black family. Arch Neurol 58: 296 – 299, 2001 - Jardin LB et al., Neurologic findings in Machado‐Joseph disease. Relation with disease duration,
subtypes, and (CAG)n. Arch Neurol 58: 899 – 904, 2001 - Maciel P et al., Improvement in the molecular diagnosis of Machado‐Joseph disease. Arch Neurol
58: 1821 – 1827, 2001 - Bryer A et al., The hereditary adult‐onset ataxias in South Africa. J Neurol Sci 216: 47‐54, 2003
- Bürk K et al., Cognitive deficits in spinocerebellar ataxia type 1, 2, and 3. J Neurol 250: 207 – 211,
2003 - Yoshizawa T et al., Magnetic resonance imaging demonstrates differential atrophy of pontine base
and tegmentum in Machado‐Joseph disease. J Neurol Sci 215: 45 – 50, 2003 - Brusco A et al., Molecular genetics of hereditary spinocerebellar ataxia. Mutation analysis of
spinocerebellar ataxia genes and CAG/CTG repeat expansion detection in 225 Italian families. Arch
Neurol 61: 727 – 733, 2004 - Gu W et al., The shortest expanded allele of the MJD1 gene in the Chinese MJD kindred with
autonomic dysfunction. Eyr Neurol 52: 107 – 111, 2004 - Kawai Y et al., Cognitive impairments in Machado‐Joseph disease. Arch Neurol 61: 1757‐1760,
2004 - Lu CS et al., The parkinsonian phenotype of spinocerebellar ataxia type 3 in a Taiwanese family.
Parkinsonism Relat Disord 10: 369 – 373, 2004 - Nandagopal R and Moorthy SGK: Dramatic levodopa responsiveness of dystonia in a sporadic case
of spinocereebellar ataxia type 3. Postgrad Med J 80: 363 – 365, 2004 - van de Warrenburg BPC et al., Peripheral nerve involvement in spinocerebellar ataxias. Arch
Neurol 61: 257 – 261, 2004 - Gould VFC: Mouse models of Machado‐Joseph Disease and other polyglutamine spinocerebellar
ataxias. Am Soc Exp Neurother 2: 480 – 483, 2005 - Juvonen V et al. The occurence of dominant spinocerebellar ataxias among 251 Finnish ataxia
patients and the role of predisponding large normal alleles in a genetically isolated population.
Acta Neurol Scand 111: 154 – 1562, 2005 - Padiath QS et al., Identification of a novel 45 repeat unstable allele associated with a disease
phenotype at the MJD1/SCA3 locus. Am J Med Genet B Neuropsychiatr 133: 124 – 126, 2005 - van de Warrenburg BPC et al., Age at onset variance analysis in spinocerebellar ataxias: a study in
a Dutch‐Frech cohort. Ann Neurol 57: 505 – 512, 2005 - Yeh T‐H et al., Autonomic dysfunction in Machado‐Joseph disease. Arch Neurol 62: 630 – 636,
2005
- Nakano KK et al: Machado disease: a hereditary ataxia in Portugenese immigrants to
Järjestötoiminta
Potilasjärjestö: Neuroliitto ry; ataksiaverkosto
Muita tietolähteitä
- The National Ataxia Foundation: www.ataxia.org
- Sveriges socialstyrelsens kunskapsdatabas om ovanliga diagnoser innehåller information om fler än 300 ovanliga sjukdomar och tillstånd.
- Tutustu myös Neuroliiton oppaaseen Helposti nieltävä – Apua nielemisen vaikeuksiin