
Spinoserebellaarinen ataksia tyyppi 4 eli SCA4
- ICD–10: G11.8, OMIM: 600223, ORPHA: 98765
- Muut sairaudesta käytetyt nimet: Hereditaarinen ataksia ja sensorinen neuropatia
Spinoserebellaarisen ataksian eli SCA4:n oireisiin kuuluvat pikkuaivo-oireiden (tasapainovaikeudet, kävely- ja raaja-ataksia sekä puheen sammaltaminen) lisäksi etenevä ääreishermoston tuntohermoihin painottuva vaurio (sensomotorinen polyneuropatia). Sairautta aiheuttava geeni on paikallistettu kromosomiin 16. SCA4 kuvattiin alun perin amerikkalaisessa suvussa vuonna 1994. Vuonna 2000 japanilaiset tutkijat raportoivat autosomisesti vallitsevasti periytyvän ataksiasairauden, jonka geenivirhe paikallistui niin ikään kromosomiin 16 samalle alueelle kuin amerikkalaisen perheen SCA4-geenimuutos. Japanilaisessa SCA4-suvussa oireet rajoittuvat pikkuaivo-oireisiin. SCA4-sairaudessa on siis todettu kaksi erilaista kliinistä oireistoa (fenotyyppiä) ja toistaiseksi näiden saman geenialueen virheistä johtuvia sairauksia pidetään keskenään alleellisina sairauksina. Sittemmin SCA4-sairautta on kuvattu eurooppalaisissa suvuissa. Eurooppalaisissa suvuissa sairastuneiden oireet ovat samankaltaisia kuin amerikkalaisella suvulla. SCA4 periytyy autosomisesti vallitsevasti eli dominantisti.
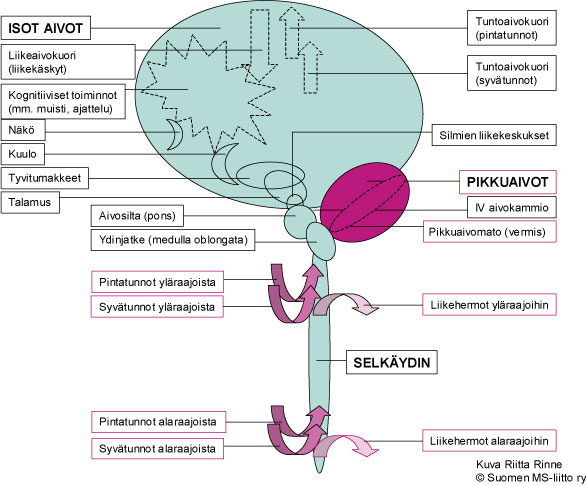
Kuva 1. Japanissa kuvatussa SCA4-perheessä oireet rajoittuivat pikkuaivo-oireisiin. Amerikkalaisessa ja saksalaisessa SCA4-suvussa todettiin pikkuaivo-oireiden lisäksi tuntohermojen vaurio. Kuvassa hermovaurioiden sijainti on merkitty punaisella.
Amerikkalaisessa ja saksalaisissa suvuissa SCA4:n oireet ovat alkaneet km. 40 vuoden iässä (39 ja 38.3 v; vaihtelu 19–66 v.) ja japanilaisissa suvuissa puolestaan 55 vuoden iässä (45–72 v.). Niin amerikkalaisessa kuin eurooppalaisissa perheissä sairastuneilla on sekä pikkuaivo-oireita että ääreishermoston toimintavaurio (Kuva 1.). SCA4 on hidaskulkuinen eikä pääsääntöisesti vaikuta sairastavien elinikään.
SCA4:n taudinkuvaan kuuluvat:
- etenevä tasapainon ja kävelyn vaikeus (kävelyataksia)
- raajojen liikehäiriöt (ataksia)
- tuntohermojen samoin kuin alempien liikehermojen vaurio
- lihasten surkastuminen (amyotrofia) ja jänneheijasteiden vaimeneminen (hyporefleksia) / sammuminen (arefleksia)
- hienomotoriikan epätarkkuus
- puheen ongelmat (dysartria)
Osalla todetaan lisäksi:
- lihasjänteyden nousua (spastisiteettia) ja heijastevilkkautta
- silmien liikehäiriöitä ei ole todettu, ei myöskään puheen- tai nielemisen ongelmia
Japanilaisissa SCA4-suvuissa oireet rajoittuivat pikkuaivojen toimintahäiriöön. Kaikilla sairastuneilla on todettu tasapainon ja kävelyn epävarmuus, raajojen koordinaatio-ongelmat, lihasjänteyden heikkeneminen (hypotonia) sekä lähes kaikilla puheen sammaltaminen (dysartria). Osalla potilaista oli silmänvärvettä (horisontaalinen nystagmus sekä värinätunnon heikkeneminen ja kantapäiden heijasteiden vaimeneminen. Osalla potilaista on todettu kuulon heikentymistä (sensorineuraalinen kuulovika). Kliininen oireisto (fenotyyppi) amerikkalaisessa ja eurooppalaisissa suvuissa poikkeaa japanilaisesta SCA4-suvusta. Alla olevassa listassa on amerikkalaisen SCA4-suvun jäsenten oireet ja löydökset.
Amerikkalaisen SCA4-suvun sairastavien oireet ja oireen esiintyvyys (%); n = 20
- Kävelyn epävarmuus (kävelyataksia) 95 %
- Raajojen koordinaatio-ongelmat (dysmetria) 95 %
- Heikentynyt kipu-, ja lämpötunnot 100 %
- Heikentynyt nivelten asento- ja tärinätunto 100 %
- Heikentynyt kosketustunto 95 %
- Jänneheijasteiden muutokset 100 %
- Akillesheijasteiden puuttuminen 100 %
- Patellaheijasteiden puuttuminen 85 %
- Kaikkien heijasteiden puuttuminen 25 %
- Puheen epäselvyys (dysartria) 50 %
- Raajojen kärkiosien lihasheikkous 20 %
- Raajojen lihasheikkous 10 %
- Ojentava jalkapohjan heijaste 20 %
- Silmien liikehäiriöt 15 %
- Pintatuntojen heikkeneminen 100 %
SCA4:n taudinkuva ja oireiden eteneminen
SCA4 alkaa tavallisimmin tasapainon epävarmuutena. Hienoinen tasapainohaitta voidaan havaita rappuja laskeutuessa tai horjahteluna käännöksissä. Vähittäisesti kävely muuttuu leveäraiteiseksi ja sivuaskeleita otetaan herkästi. Kävelyn ongelmat korostuvat hämärässä ja pimeässä.
Käsinäppäryys heikkenee. Pienten esineiden käsittely tuottaa ongelmia ja käden osumatarkkuus huononee. Monimutkaisia liikeliiketaitoja ei opita ja jo opittujen taitoja menetetään. Heikentynyt nivelten asentotunto edellyttää katsekontrollia tarkoissa ja sairauden edetessä myös tavanomaisissa, varsin yksinkertaisissa suorituksissa.
Amerikkalaisessa suvussa kaikilla todettiin vaikea-asteista ääreishermoston toimintaheikkoutta: tunnon alenemaa sekä lihasten jänteyden heikkenemistä ja voimattomuutta. Ajan myötä lihakset kuihtuivat. Japanilaisissa perheissä ääreishermostomuutokset eivät kuuluneet taudinkuvaan, sen sijaan lähes kaikilla (92.6 %:lla) todettiin puheongelmia, joita ei puolestaan amerikkalaisella suvulla todettu.
SCA4:n periytyminen
SCA4 periytyy autosomisesti vallitsevasti eli autosomissa dominantisti (AD). Sairautta aiheuttava geenivirhe tunnistettiin kromosomissa 16(q22) vuonna 1994. Vuonna 2000 japanilaisissa SCA-perheissä tunnistettiin saman geenialueen (16q22.1) virheestä johtuvan autosomisesti vallitsevasti periytyvän SCA-sairauden. Kromosomi 16q22 geenimuutos on todettu sittemmin eurooppalaisilla ja myös muutamilla skandinaavisilla suvuilla. Kaikilta tutkituilta japanilaissuvuilta on löydetty pistemutaatio Puratrofiini-1 valkuaisainetta (Purkinje cell atrophy associated protein-1) ohjaavassa geenissä (Kuva 2.)
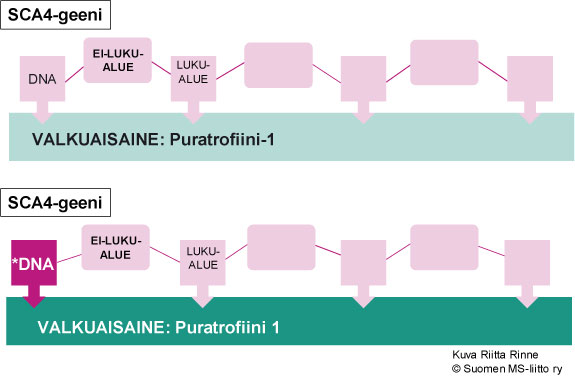
Kuva 2. Japanilaisilla SCA4-suvuilla on todettu geenimutaatio Puratrofiini-1 valkuaisainetta ohjaavassa geenissä kromosomissa 16q22.1.
Antisipaatiolla tarkoitetaan sairauden puhkeamista keskimäärin varhemmin seuraavassa sukupolvessa. Japanilaisilla perheillä todettiin vähäinen SCA4:n antisipaatio, mikä merkitsi seuraavan sukupolven oireiden puhkeamista km. 5 vuotta aikaisemmin. Amerikkalaisen suvun antisipaatio IV:ssä ja V:ssä sukupolvessa oli samoin 5 vuotta: IV:n sukupolven oireet alkoivat km. 41.9 vuoden iässä ja V:n sukupolven oireet keskimäärin 36,7 vuoden iässä.
Perinnöllisyysneuvonta on aiheellista järjestää aina epäiltäessä SCA-sairautta sekä sairastuneelle että perheenjäsenille. Neuvonnassa annetaan tietoa niin sairauden laadusta kuin myös sen perinnöllisyydestä ja sairastumisen riskistä. Neuvonnassa voi keskustella myös ennakoivan geenitestin (prediktiivinen geenitesti) mahdollisuudesta.
SCA4:n diagnoosi ja erotusdiagnoosi
SCA4-sairauden diagnoosiin ja erotusdiagnoosiin tarvitaan neurologinen tutkimus sekä sen perusteella arvioitavat laboratorio-, kuvantamis- ja muut hermotoimintojen tutkimukset. Muiden erikoisalojen konsultaatiot ovat perusteltuja. Perinnöllisyyslääkärin konsultaatio ja neuvonta sairastuneelle ja sukulaisille on aina tarpeen.
Laboratoriotutkimukset verinäytteestä ovat normaalit, samoin selkäydinnestenäyte (likvor-tutkimus). Aivojen magneettitutkimuksessa nähdään atrofiaa pikkuaivomadossa (vermiksessä) ja pikkuaivokupoleissa (hemisfääreissä). Japanilaisilla perheillä ei todettu muutoksia ääreishermoston tutkimuksissa, kun taas amerikkalaisessa suvussa aksonaalinen tuntohermovaurio oli aina todettavissa. Japanilaisissa suvuissa neuro-ontologisissa tutkimuksissa havaittiin kuulovaikeutta lähes joka toisella tutkituista (42,9%:lla) (sensorineuraalinen kuulovika). Aivojen magneettitutkimuksessa nähdään atrofiaa pikkuaivomadossa (vermiksessä) ja pikkuaivokupoleissa (hemisfääreissä).
Erotusdiagnostiikassa tulevat kyseeseen ennen kaikkea muut SCA-sairaudet (ADCA tyypit I ja III). Erotusdiagnostiikassa tulevat poissuljettaviksi ääreishermoston sairaudet, erityisesti autosomissa dominantisti periytyvät sensomotoriset neuropatiat (HMSN-sairaudet) ja Refsumin tauti.
SCA4:n esiintyminen
SCA4-sairaus on erittäin harvinainen SCA-sairaus. Toistaiseksi SCA4-sukuja on kuvattu Amerikassa Utahin osavaltiossa, Japanissa ja Saksassa.
SCA4:n aiheuttamat hermovauriot
SCA4:n neuropatologisina muutoksina todetaan sekä pikkuaivojen että aivorungon myeliinikato (demyelinisaatio) ja pikkuaivojen ja aivorungon hermosolukato (atrofia). Atrofiamuutoksia todettan myös tyvitumakkeissa (substantia nigra) sekä keskiaivoissa (ventraalinen tegmentum), aivorungon raphe-, pons- ja olivatumakkeissa, aivohermoissa (IV, V, VII, VIII, X, XII) ja niiden tumakkeissa todettiin rappeutumista, samoin selkäytimen tumakkeissa. Pikkuaivojen Purkinjen soluissa sekä fastigii-tumakkeissa on hermosolukatoa.
SCA4:n hoito ja kuntoutus
Sairauden hoitoon ei ole käytettävissä oireiden etenemistä hillitsevää tai sairautta parantavaa lääkehoitoa. Vitamiinilisiä on suositeltu – erityisesti silloin, kun tavanomaisesta ravinnosta niitä ei todennäköisesti riittävästi kerry. Perinnöllisyysneuvonta on oleellinen osa SCA4-sairauden hoitoa. Neuvontaa tarvitsevat sairastuneen lisäksi myös muut perheenjäsenet.
SCA4-sairaudessa kuntoutustoimet on syytä aina arvioida yksilöllisesti samoin kuin tarvittavat palvelut. Sairaus etenee yleensä hitaasti, mutta saattaa aiheuttaa muutostarvetta niin työ- kuin arkielämässä.
SCA4:n perustutkimus
SCA4:n geenimutaatio on kromosomissa 16 (q22). Geeni määrää aina tietyn yhden valkuaisaineen eli proteiinin laadun. Japanilaisissa suvuissa varmistunut SCA4-sairautta aiheuttava geeni ohjaa Puratrofiini-1 valkuaisaineen tuottamista. Puratrofiini-1 valkuaisaine sisältää mm. spektriini-jakson. Puratrofiini-1 proteiini on liitetty kiinteästi Purkinjen solujen toimintaan. Se osallistuu soluviestintään sekä Golgin laitteen toimintaan. Seurauksena geenin toimimattomuudesta on todettu solunsisäisiä kertymiä, jotka eivät ole PolyQ-ataksioissa todettujen kertymien kaltaisia. Toistaiseksi ei tiedetä, onko amerikkalaisessa ja eurooppalaisissa suvuissa kuvattu SCA4-sairaus Puratrofiini-1:n toimintahäiriöstä johtuva. Todennäköistä on, että eri maanosissa kuvatut SCA4-suvut ovat geneettisesti alleelisia.
Kirjoittaja
Riitta Rinne, LL, neurologian erikoislääkäri, Maskun neurologinen kuntoutuskeskus
Julkaistu 14.10.2002. Päivitetty 10.10.2006
- Kirjallisuusviitteet
- Gardner K et al., Autosomal dominant spinocerebellar ataxia: clinical description of a distinct
hereditary ataxia and genetic localization to chromosome 16 (SCA4) in a Utah Kindred. Neurology
44: A361, 1994 - Flanigan K et al., Autosomal dominant spinocerebellar ataxia with sensory axonal neuropathy
(SCA4): clinical description and genetic localization to chromosome 16q22.1. Am J Hum Genet 59:
392 – 399, 1996 - Kaakkola S ja Rinne R: Ataksiat ja niiden erotusdiagnostiikka (katsaus). Duodecim 113: 1773 –
1782, 1997 - Nachmanoff DB et al., Hereditary ataxia with sensory neuronopathy: Biemond´s ataxia. Neurology
48: 273 – 275, 1997 - Fu Y‐H et al., Spinocerebellar Ataxia Type 4. Kirjassa: Handbook of Ataxia Disorders, ss. 426 ‐ 433.
Ed. Klockgether T, Marcel Dekker Inc., New York‐Basel, 2000 - Nagaoka U et al., A gene on SCA4 locus causes dominantly inherited pure cerebellar ataxia.
Neurology 54: 1971 – 1975, 2000 - Hirano R et al., Fine mapping of 16q‐linked autosomal dominant ceebellar ataxia type III in
Japanese families. Neurogenetics 5: 215 – 221, 2004 - Hallenbroich Y et al., Spinocerebellar ataxia type 4. Investigation of 34 candidate genes. J Neurol
252: 1472 – 1475, 2005 - Hallenbroich Y et al., Spinocerebellar ataxia type 4 (SCA4): Initial pathoanatomical study reveals
widespared cerebellar and brainstem degeneration. J Neural Transm, 2005 - Ishigawa K et al., An autosomal dominant cerebeöllar ataxia linked to chromosome 16q22.1 is
associated with a single‐nucleotide substitution in the %´untranslated region of the gene encoding
a protein with spectrin repeat and Rho guanine‐nucleotide exchange‐factor domains. Am J Hum
Genet 77: 280 – 296, 2005
- Gardner K et al., Autosomal dominant spinocerebellar ataxia: clinical description of a distinct
Järjestötoiminta
Potilasjärjestö: Neuroliitto ry; Ataksiaverkosto
Muut tietolähteet
- The National Ataxia Foundation: www.ataxia.org
- Sveriges socialstyrelsens kunskapsdatabas om ovanliga diagnoser innehåller information om fler än 300 ovanliga sjukdomar och tillstånd.